

Oxidative Stress Attenuates Lipid Synthesis and Increases Mitochondrial Fatty Acid Oxidation in Hepatoma Cells Infected with Hepatitis C Virus
- PMID: 26627833
- PMCID: PMC4722472
- DOI: 10.1074/jbc.M115.674861
Oxidative Stress Attenuates Lipid Synthesis and Increases Mitochondrial Fatty Acid Oxidation in Hepatoma Cells Infected with Hepatitis C Virus
Abstract
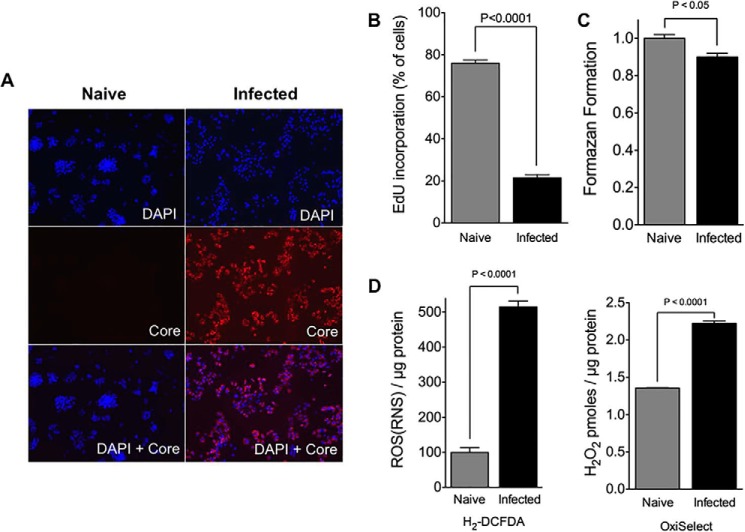
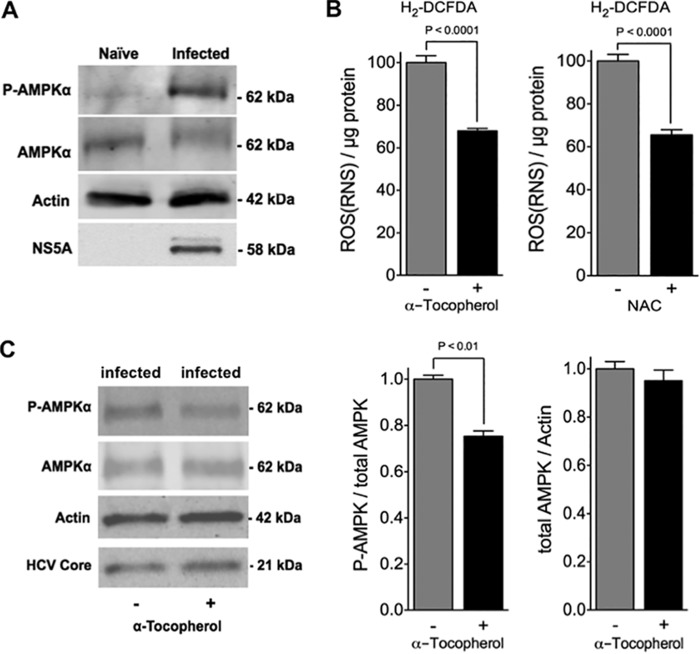
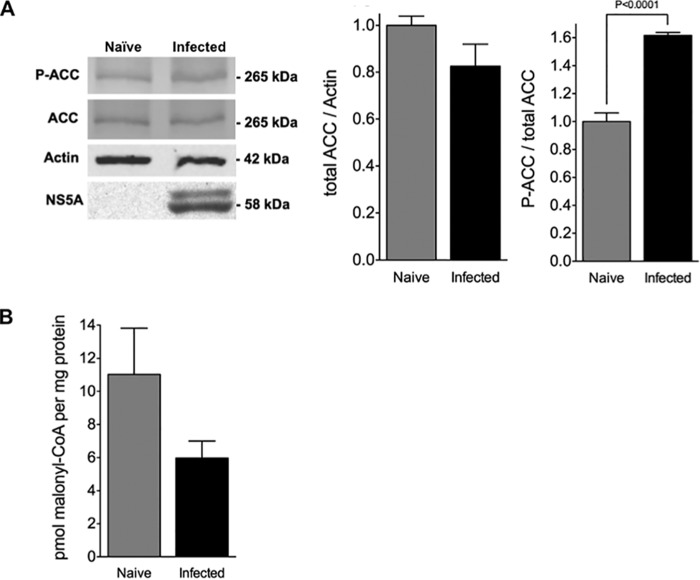
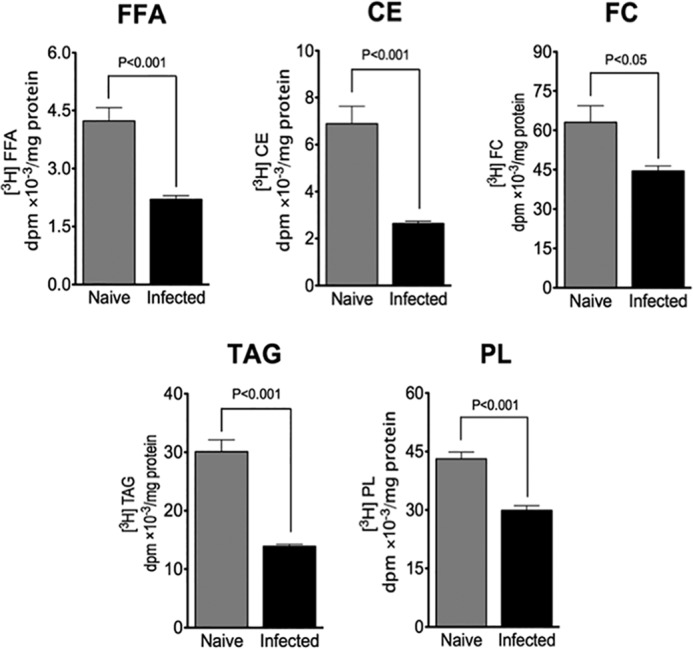
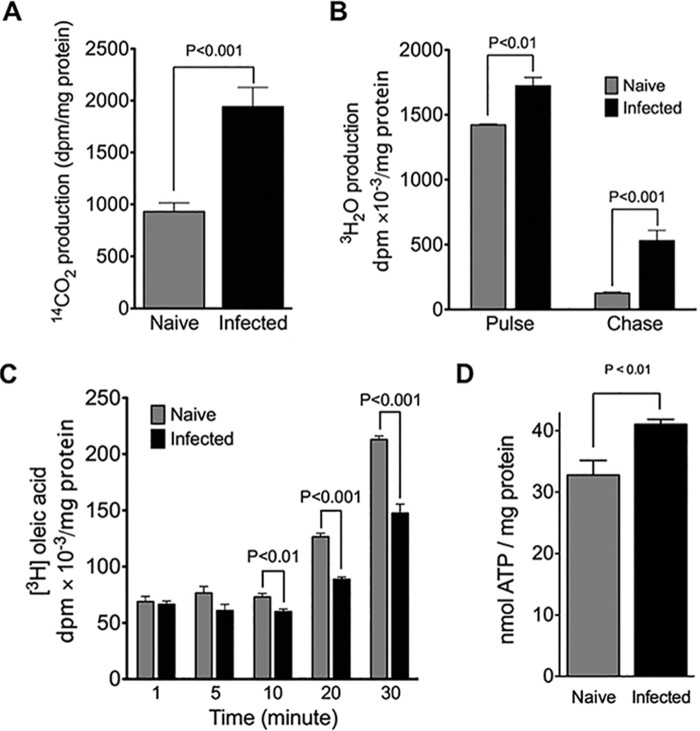
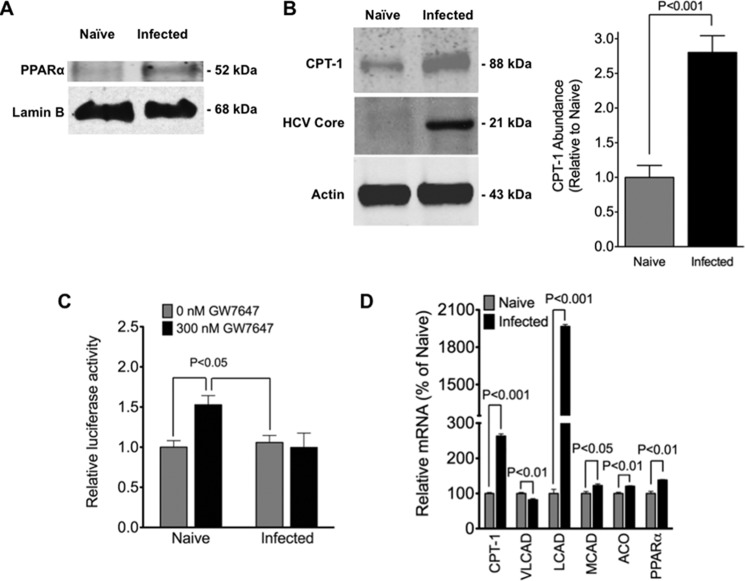
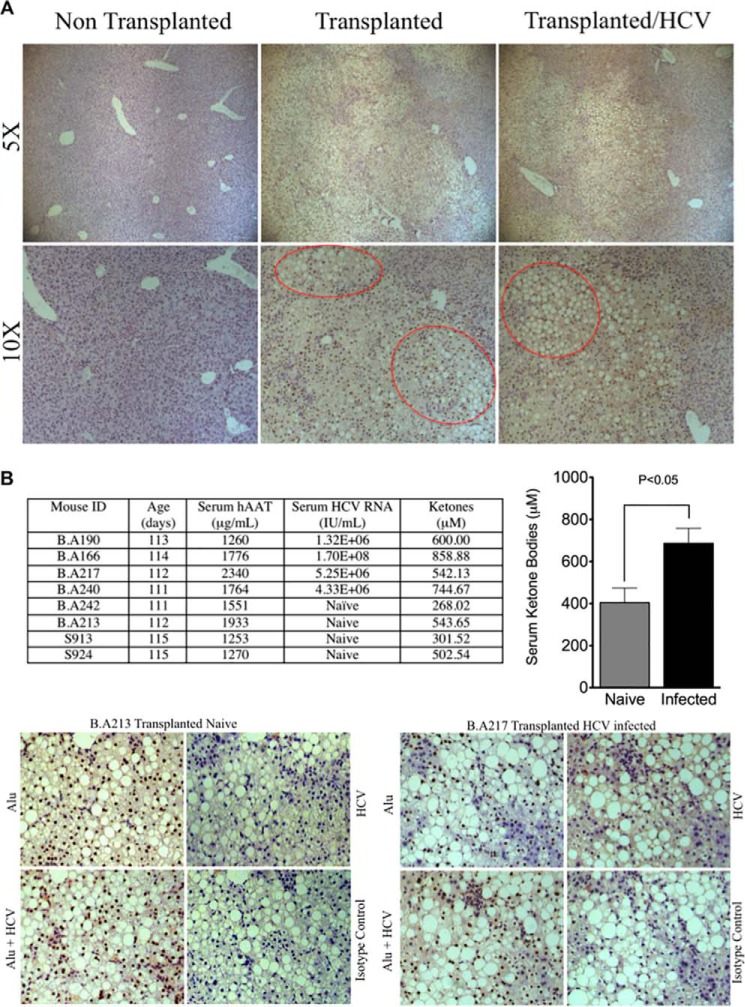
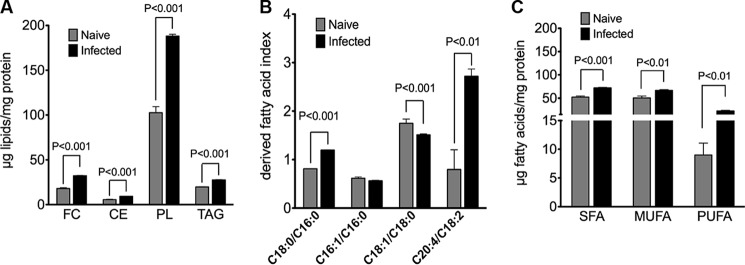
Cytopathic effects are currently believed to contribute to hepatitis C virus (HCV)-induced liver injury and are readily observed in Huh7.5 cells infected with the JFH-1 HCV strain, manifesting as apoptosis highly correlated with growth arrest. Reactive oxygen species, which are induced by HCV infection, have recently emerged as activators of AMP-activated protein kinase. The net effect is ATP conservation via on/off switching of metabolic pathways that produce/consume ATP. Depending on the scenario, this can have either pro-survival or pro-apoptotic effects. We demonstrate reactive oxygen species-mediated activation of AMP-activated kinase in Huh7.5 cells during HCV (JFH-1)-induced growth arrest. Metabolic labeling experiments provided direct evidence that lipid synthesis is attenuated, and β-oxidation is enhanced in these cells. A striking increase in nuclear peroxisome proliferator-activated receptor α, which plays a dominant role in the expression of β-oxidation genes after ligand-induced activation, was also observed, and we provide evidence that peroxisome proliferator-activated receptor α is constitutively activated in these cells. The combination of attenuated lipid synthesis and enhanced β-oxidation is not conducive to lipid accumulation, yet cellular lipids still accumulated during this stage of infection. Notably, the serum in the culture media was the only available source for polyunsaturated fatty acids, which were elevated (2-fold) in the infected cells, implicating altered lipid import/export pathways in these cells. This study also provided the first in vivo evidence for enhanced β-oxidation during HCV infection because HCV-infected SCID/Alb-uPA mice accumulated higher plasma ketones while fasting than did control mice. Overall, this study highlights the reprogramming of hepatocellular lipid metabolism and bioenergetics during HCV infection, which are predicted to impact both the HCV life cycle and pathogenesis.
Keywords: AMP-activated kinase (AMPK); hepatitis C virus (HCV); lipogenesis; oxidative stress; β-oxidation.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Hepatocarcinogenesis in hepatitis C: HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species.Oncology. 2011;81 Suppl 1:11-7. doi: 10.1159/000333253. Epub 2011 Dec 22. Oncology. 2011. PMID: 22212930
-
Lipid metabolism and liver disease in hepatitis C viral infection.Oncology. 2010 Jul;78 Suppl 1:24-30. doi: 10.1159/000315226. Epub 2010 Jul 8. Oncology. 2010. PMID: 20616580
-
Pathogenesis and significance of hepatitis C virus steatosis: an update on survival strategy of a successful pathogen.World J Gastroenterol. 2014 Jun 21;20(23):7089-103. doi: 10.3748/wjg.v20.i23.7089. World J Gastroenterol. 2014. PMID: 24966582 Free PMC article. Review.
-
Transforming Growth Factor β Acts as a Regulatory Molecule for Lipogenic Pathways among Hepatitis C Virus Genotype-Specific Infections.J Virol. 2019 Aug 28;93(18):e00811-19. doi: 10.1128/JVI.00811-19. Print 2019 Sep 15. J Virol. 2019. PMID: 31243135 Free PMC article.
-
[Possible mechanisms of hepatitis C virus-related hepatocarcinogenesis].Nihon Rinsho. 2004 Nov;62(11):2137-44. Nihon Rinsho. 2004. PMID: 15552900 Review. Japanese.
Cited by
-
Lipid Catabolism and ROS in Cancer: A Bidirectional Liaison.Cancers (Basel). 2021 Oct 31;13(21):5484. doi: 10.3390/cancers13215484. Cancers (Basel). 2021. PMID: 34771647 Free PMC article. Review.
-
2-Bromopalmitate depletes lipid droplets to inhibit viral replication.J Virol. 2024 Apr 16;98(4):e0017124. doi: 10.1128/jvi.00171-24. Epub 2024 Mar 15. J Virol. 2024. PMID: 38488361 Free PMC article.
-
Mitochondrial Proteins as Metabolic Biomarkers and Sites for Therapeutic Intervention in Primary and Metastatic Cancers.Mini Rev Med Chem. 2024;24(12):1187-1202. doi: 10.2174/0113895575254320231030051124. Mini Rev Med Chem. 2024. PMID: 39004839 Review.
-
Transcriptome and miRNome Analysis Provide New Insight Into Host Lipid Accumulation, Innate Immunity, and Viral Persistence in Hepatitis C Virus Infection in vitro.Front Microbiol. 2020 Sep 30;11:535673. doi: 10.3389/fmicb.2020.535673. eCollection 2020. Front Microbiol. 2020. PMID: 33101221 Free PMC article.
-
Iron, Oxidative Stress, and Δ9 Stearoyl-CoenzymeA Desaturase Index (C16:1/C16:0): An Analysis Applying the National Health and Nutrition Examination Survey 2003-04.Curr Dev Nutr. 2017 Nov 15;2(1):1-8. doi: 10.1093/cdn/nzx001. eCollection 2018 Jan. Curr Dev Nutr. 2017. PMID: 29955721 Free PMC article.
References
-
- Alter M. J., Margolis H. S., Krawczynski K., Judson F. N., Mares A., Alexander W. J., Hu P. Y., Miller J. K., Gerber M. A., and Sampliner R. E. (1992) The natural history of community-acquired hepatitis C in the United States. The Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. N. Engl. J. Med. 327, 1899–1905 - PubMed
-
- Nelson D. R. (2001) The immunopathogenesis of hepatitis C virus infection. Clin. Liver Dis. 5, 931–953 - PubMed
-
- Mengshol J. A., Golden-Mason L., and Rosen H. R. (2007) Mechanisms of disease: HCV-induced liver injury. Nat. Clin. Pract. Gastroenterol. Hepatol. 4, 622–634 - PubMed
-
- Thomas D. L., and Seeff L. B. (2005) Natural history of hepatitis C. Clin. Liver Dis. 9, 383–398 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
