

Wham: Identifying Structural Variants of Biological Consequence
- PMID: 26625158
- PMCID: PMC4666669
- DOI: 10.1371/journal.pcbi.1004572
Wham: Identifying Structural Variants of Biological Consequence
Abstract
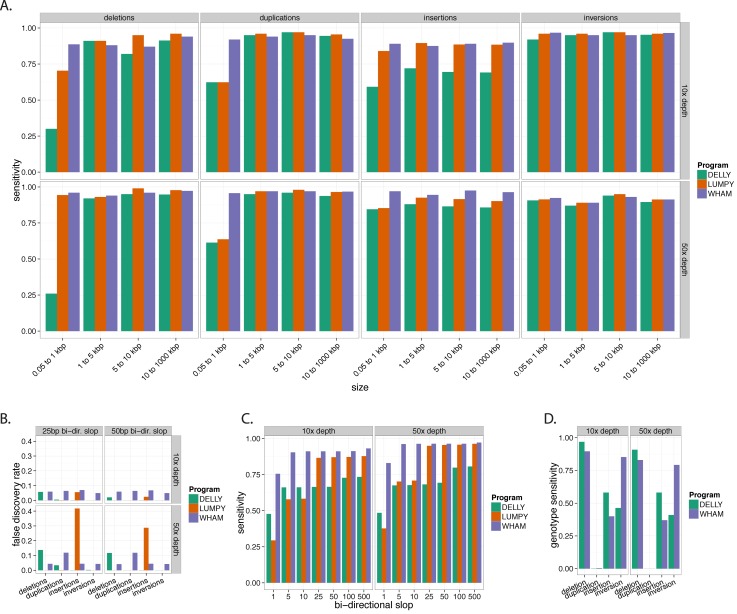
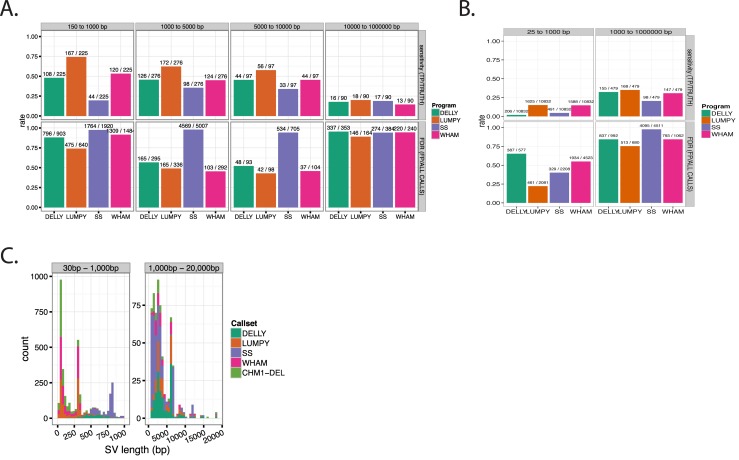
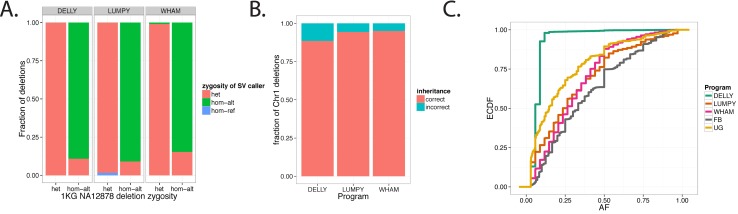
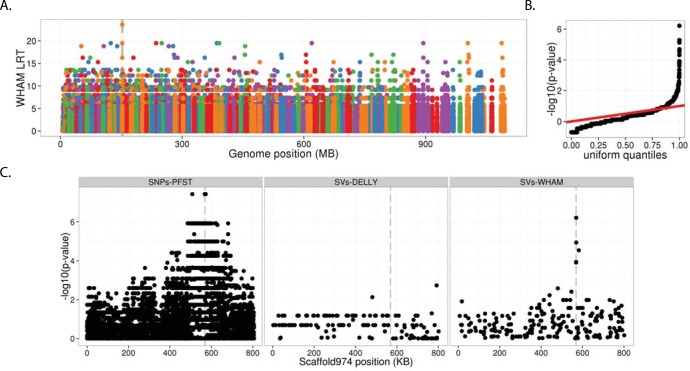
Existing methods for identifying structural variants (SVs) from short read datasets are inaccurate. This complicates disease-gene identification and efforts to understand the consequences of genetic variation. In response, we have created Wham (Whole-genome Alignment Metrics) to provide a single, integrated framework for both structural variant calling and association testing, thereby bypassing many of the difficulties that currently frustrate attempts to employ SVs in association testing. Here we describe Wham, benchmark it against three other widely used SV identification tools-Lumpy, Delly and SoftSearch-and demonstrate Wham's ability to identify and associate SVs with phenotypes using data from humans, domestic pigeons, and vaccinia virus. Wham and all associated software are covered under the MIT License and can be freely downloaded from github (https://github.com/zeeev/wham), with documentation on a wiki (http://zeeev.github.io/wham/). For community support please post questions to https://www.biostars.org/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
RAPTR-SV: a hybrid method for the detection of structural variants.Bioinformatics. 2015 Jul 1;31(13):2084-90. doi: 10.1093/bioinformatics/btv086. Epub 2015 Feb 16. Bioinformatics. 2015. PMID: 25686638
-
DELLY: structural variant discovery by integrated paired-end and split-read analysis.Bioinformatics. 2012 Sep 15;28(18):i333-i339. doi: 10.1093/bioinformatics/bts378. Bioinformatics. 2012. PMID: 22962449 Free PMC article.
-
Comprehensive evaluation and guidance of structural variation detection tools in chicken whole genome sequence data.BMC Genomics. 2024 Oct 16;25(1):970. doi: 10.1186/s12864-024-10875-1. BMC Genomics. 2024. PMID: 39415108 Free PMC article.
-
Geographic distribution and adaptive significance of genomic structural variants: an anthropological genetics perspective.Hum Biol. 2014 Fall;86(4):260-75. doi: 10.13110/humanbiology.86.4.0260. Hum Biol. 2014. PMID: 25959693 Review.
-
A decade of structural variants: description, history and methods to detect structural variation.Brief Funct Genomics. 2015 Sep;14(5):305-14. doi: 10.1093/bfgp/elv014. Epub 2015 Apr 15. Brief Funct Genomics. 2015. PMID: 25877305 Review.
Cited by
-
Whole-genome sequencing identifies novel genes for autism in Chinese trios.Sci China Life Sci. 2024 Nov;67(11):2368-2381. doi: 10.1007/s11427-023-2564-8. Epub 2024 Aug 7. Sci China Life Sci. 2024. PMID: 39126614
-
SV-plaudit: A cloud-based framework for manually curating thousands of structural variants.Gigascience. 2018 Jul 1;7(7):giy064. doi: 10.1093/gigascience/giy064. Gigascience. 2018. PMID: 29860504 Free PMC article.
-
Next-Generation Sequencing Technology: Current Trends and Advancements.Biology (Basel). 2023 Jul 13;12(7):997. doi: 10.3390/biology12070997. Biology (Basel). 2023. PMID: 37508427 Free PMC article. Review.
-
Comparative whole-genome sequence analysis of Mycobacterium tuberculosis isolated from tuberculous meningitis and pulmonary tuberculosis patients.Sci Rep. 2018 Mar 20;8(1):4910. doi: 10.1038/s41598-018-23337-y. Sci Rep. 2018. PMID: 29559684 Free PMC article.
-
Wide spectrum and high frequency of genomic structural variation, including transposable elements, in large double-stranded DNA viruses.Virus Evol. 2020 Jan 27;6(1):vez060. doi: 10.1093/ve/vez060. eCollection 2020 Jan. Virus Evol. 2020. PMID: 32002191 Free PMC article.
References
-
- McCarroll S, Altshuler DM (2007) Copy-number variation and association studies of human disease. Nat Genet 39: S37–S42. - PubMed
Publication types
MeSH terms
Grants and funding
- F32GM103077/GM/NIGMS NIH HHS/United States
- F32 GM103077/GM/NIGMS NIH HHS/United States
- R01 GM104390/GM/NIGMS NIH HHS/United States
- T32 HD007491/HD/NICHD NIH HHS/United States
- R01GM114514/GM/NIGMS NIH HHS/United States
- R01 GM115996/GM/NIGMS NIH HHS/United States
- T32 GM007464/GM/NIGMS NIH HHS/United States
- R01GM115996/GM/NIGMS NIH HHS/United States
- T32GM07464/GM/NIGMS NIH HHS/United States
- T32HD07491/HD/NICHD NIH HHS/United States
- R01 GM114514/GM/NIGMS NIH HHS/United States
- TL1 TR001066/TR/NCATS NIH HHS/United States
- T32 AI055434/AI/NIAID NIH HHS/United States
- R01HG005969/HG/NHGRI NIH HHS/United States
- T32AI055434/AI/NIAID NIH HHS/United States
- UL1 TR001067/TR/NCATS NIH HHS/United States
- 1TL1TR001066/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
