

Ischemic Preconditioning in White Matter: Magnitude and Mechanism
- PMID: 26609155
- PMCID: PMC4659824
- DOI: 10.1523/JNEUROSCI.2544-15.2015
Ischemic Preconditioning in White Matter: Magnitude and Mechanism
Abstract
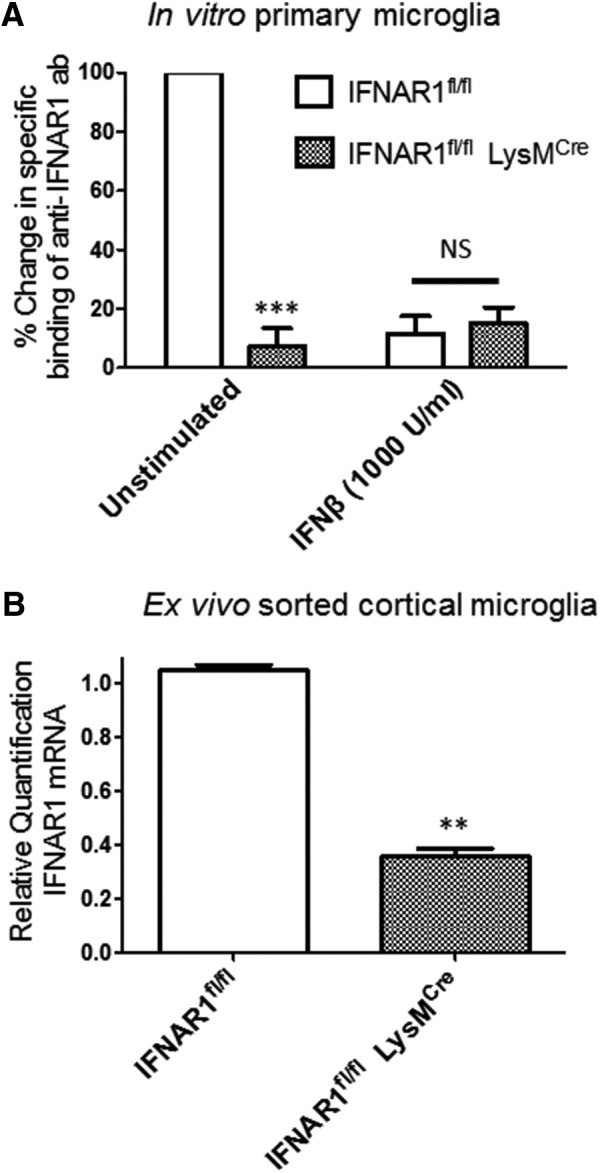
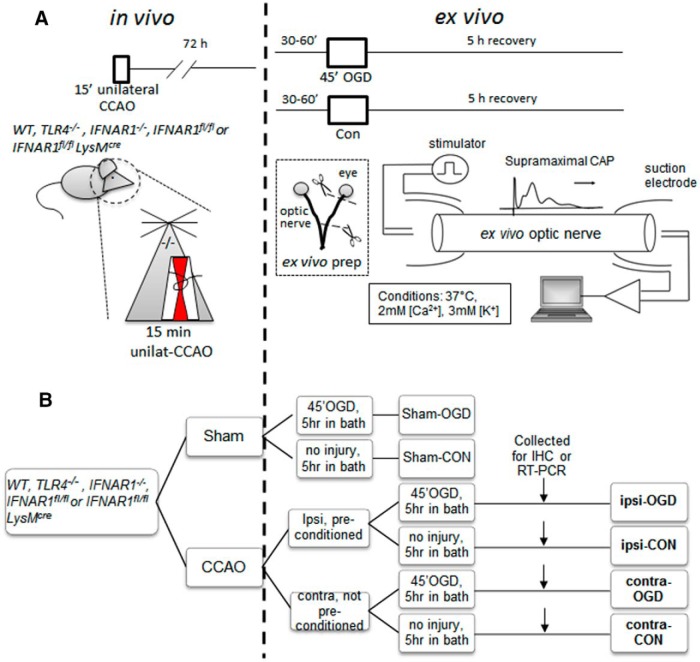
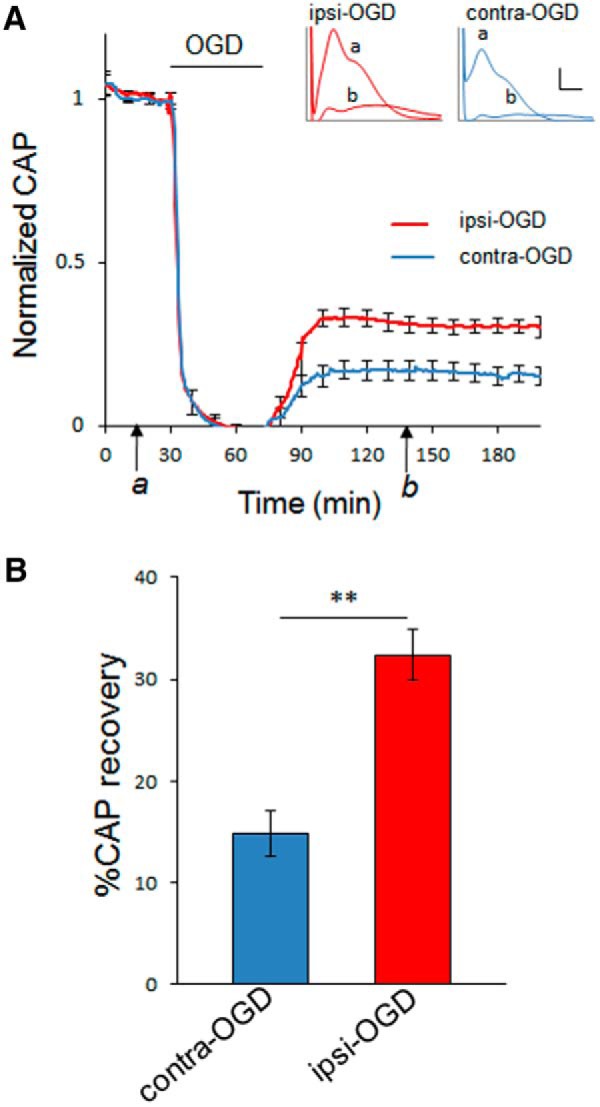
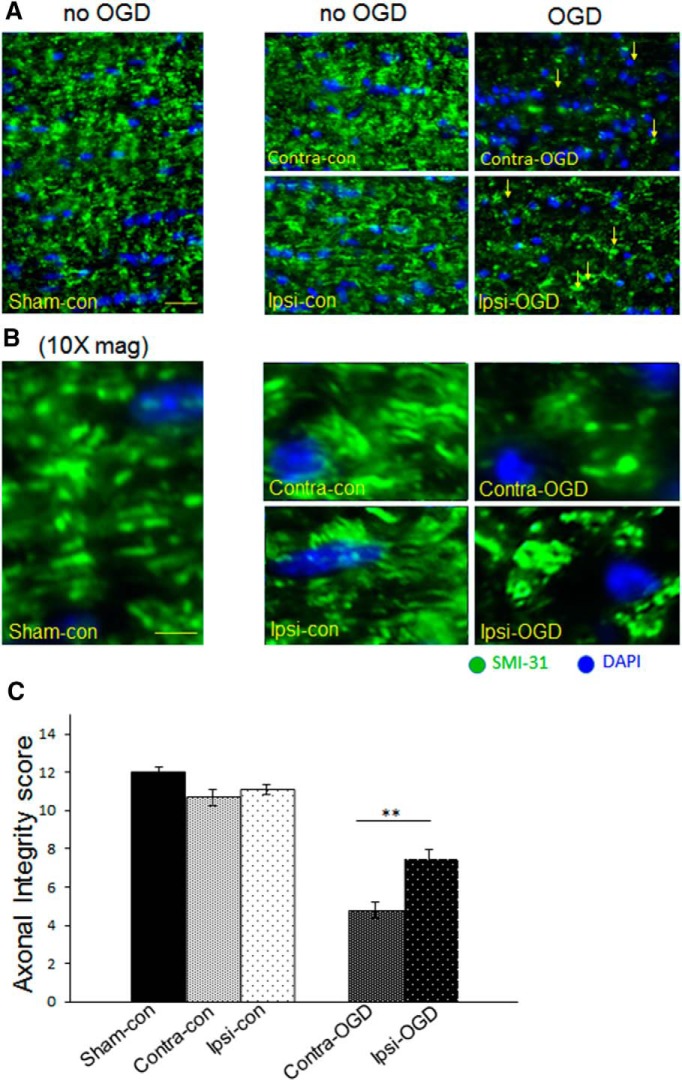
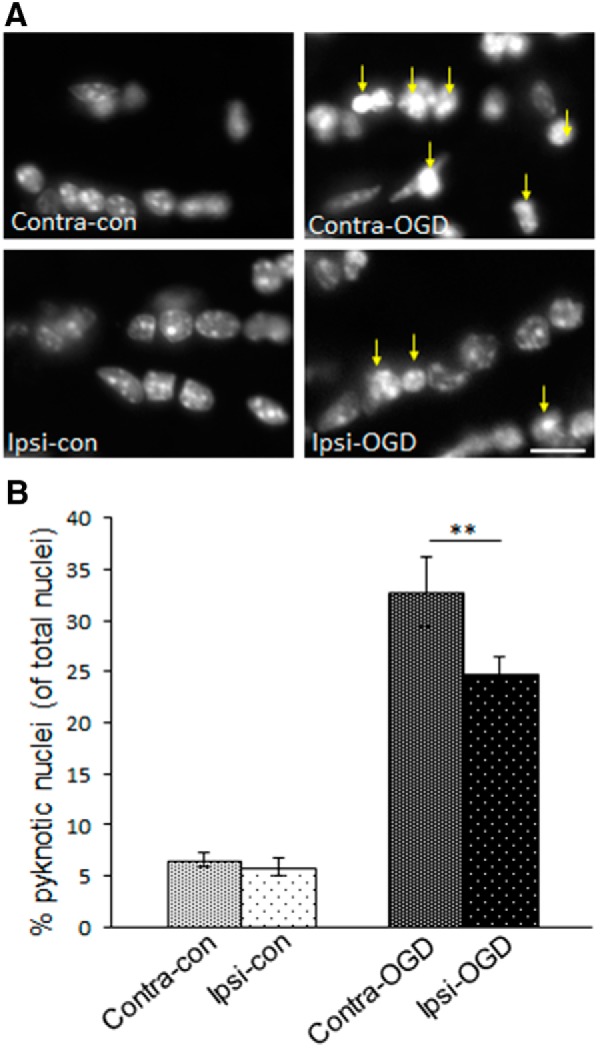
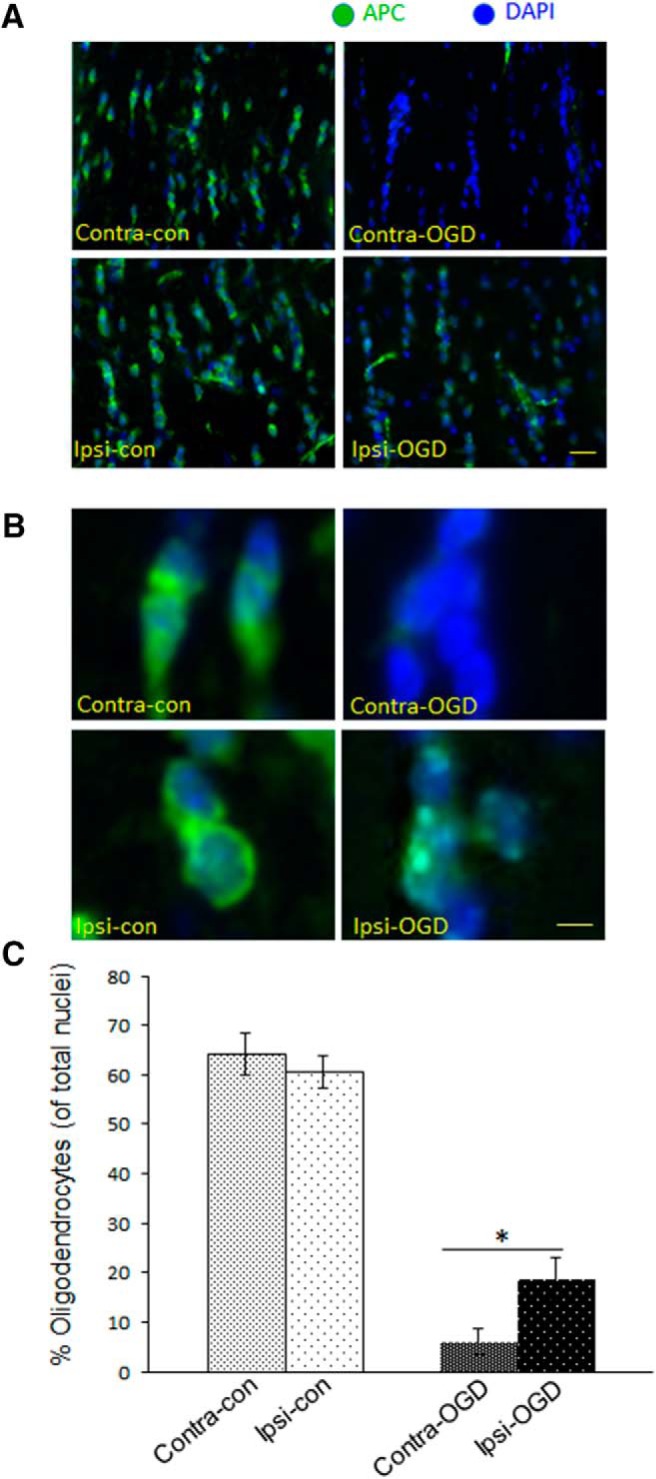
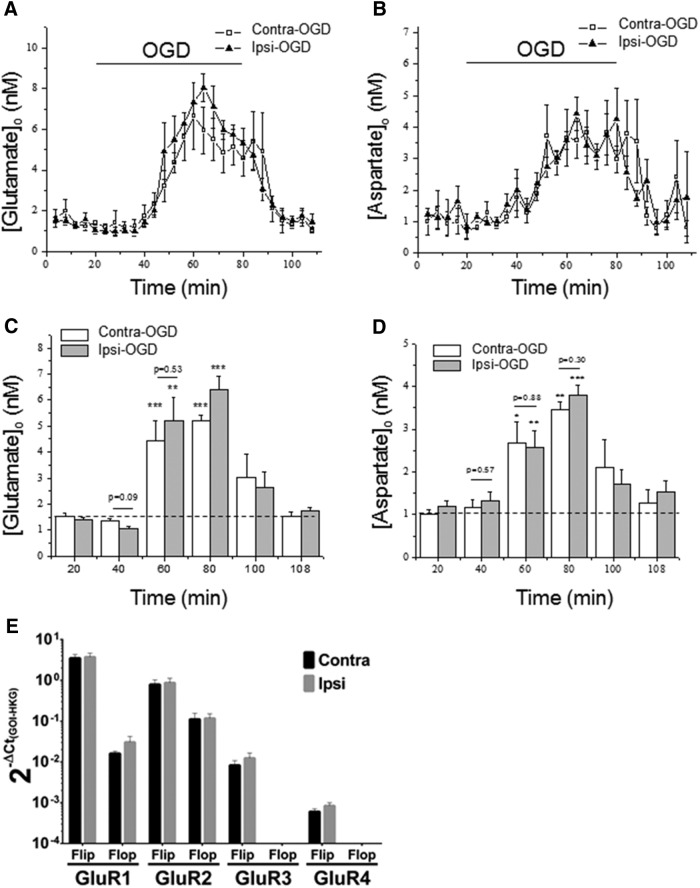
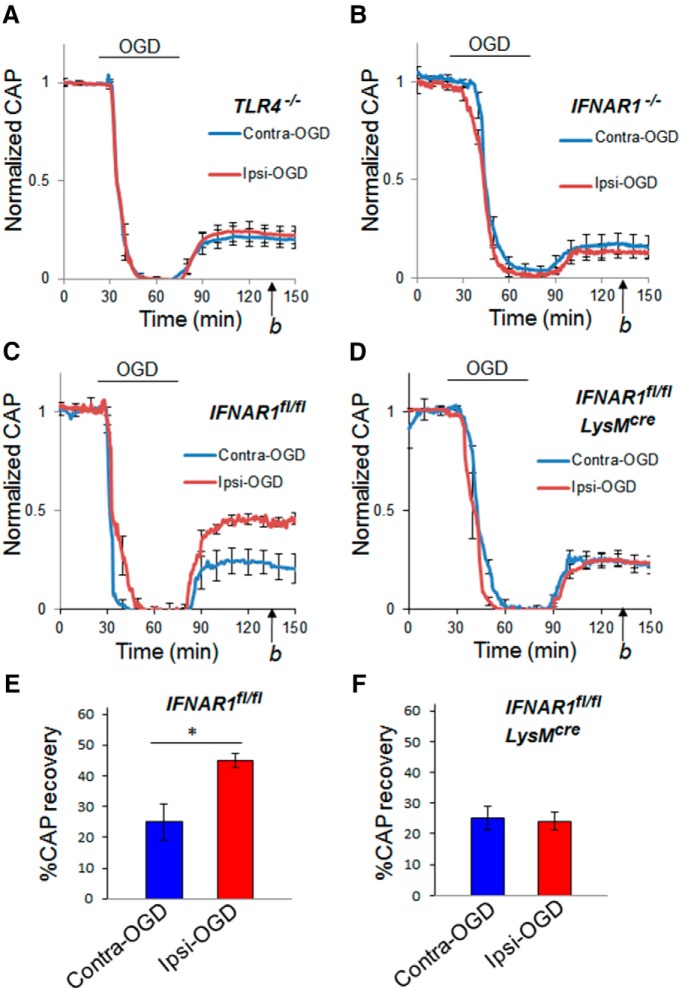
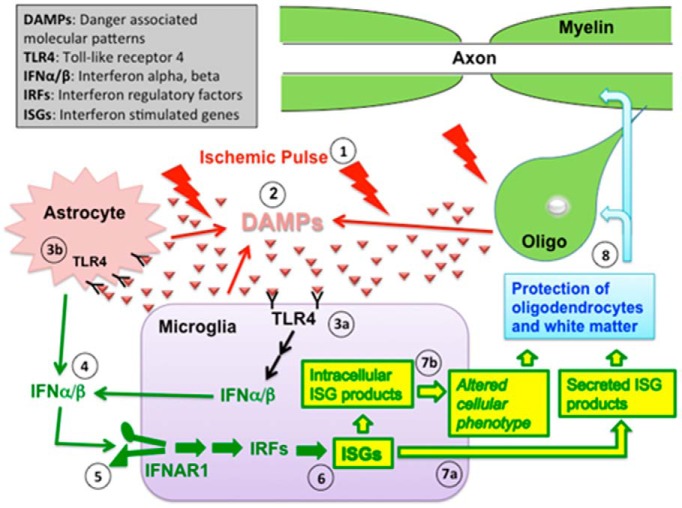
Ischemic preconditioning (IPC) is a robust neuroprotective phenomenon whereby brief ischemic exposure confers tolerance to a subsequent ischemic challenge. IPC has not been studied selectively in CNS white matter (WM), although stroke frequently involves WM. We determined whether IPC is present in WM and, if so, its mechanism. We delivered a brief in vivo preconditioning ischemic insult (unilateral common carotid artery ligation) to 12- to 14-week-old mice and determined WM ischemic vulnerability [oxygen-glucose deprivation (OGD)] 72 h later, using acutely isolated optic nerves (CNS WM tracts) from the preconditioned (ipsilateral) and control (contralateral) hemispheres. Functional and structural recovery was assessed by quantitative measurement of compound action potentials (CAPs) and immunofluorescent microscopy. Preconditioned mouse optic nerves (MONs) showed better functional recovery after OGD than the non-preconditioned MONs (31 ± 3 vs 17 ± 3% normalized CAP area, p < 0.01). Preconditioned MONs also showed improved axon integrity and reduced oligodendrocyte injury compared with non-preconditioned MONs. Toll-like receptor-4 (TLR4) and type 1 interferon receptor (IFNAR1), key receptors in innate immune response, are implicated in gray matter preconditioning. Strikingly, IPC-mediated WM protection was abolished in both TLR4(-/-) and IFNAR1(-/-) mice. In addition, IPC-mediated protection in WM was also abolished in IFNAR1(fl/fl) LysM(cre), but not in IFNAR1(fl/fl) control, mice. These findings demonstrated for the first time that IPC was robust in WM, the phenomenon being intrinsic to WM itself. Furthermore, WM IPC was dependent on innate immune cell signaling pathways. Finally, these data demonstrated that microglial-specific expression of IFNAR1 plays an indispensable role in WM IPC.
Significance statement: Ischemic preconditioning (IPC) has been studied predominantly in gray matter, but stroke in humans frequently involves white matter (WM) as well. Here we describe a novel, combined in vivo/ex vivo mouse model to determine whether IPC occurs in WM. It does. Using genetically altered mice, we identified two innate immune cell receptors, Toll-like receptor 4 and type 1 interferon receptor (IFNAR1), that are required for IPC-mediated protection in WM. Furthermore, using microglia-targeted IFNAR1 knockdown, we demonstrate that interferon signaling specifically in microglia is essential for this protection. The discovery of IPC as an intrinsic capability of WM is novel and important. This is also the first in vivo demonstration that cell-type-specific expression of an individual gene plays an indispensable role in IPC-mediated protection.
Keywords: interferon; ischemic preconditioning; microglia; toll-like receptor-4; white matter.
Copyright © 2015 the authors 0270-6474/15/3515599-13$15.00/0.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Microglial depletion abolishes ischemic preconditioning in white matter.Glia. 2022 Apr;70(4):661-674. doi: 10.1002/glia.24132. Epub 2021 Dec 23. Glia. 2022. PMID: 34939240 Free PMC article.
-
Ischemia/Reperfusion Induces Interferon-Stimulated Gene Expression in Microglia.J Neurosci. 2017 Aug 23;37(34):8292-8308. doi: 10.1523/JNEUROSCI.0725-17.2017. Epub 2017 Jul 26. J Neurosci. 2017. PMID: 28747383 Free PMC article.
-
Astrocytic Toll-like receptor 3 is associated with ischemic preconditioning-induced protection against brain ischemia in rodents.PLoS One. 2014 Jun 10;9(6):e99526. doi: 10.1371/journal.pone.0099526. eCollection 2014. PLoS One. 2014. PMID: 24914679 Free PMC article.
-
The role of microglia in ischemic preconditioning.Glia. 2020 Mar;68(3):455-471. doi: 10.1002/glia.23695. Epub 2019 Aug 6. Glia. 2020. PMID: 31386233 Free PMC article. Review.
-
Neuroimmune Response in Ischemic Preconditioning.Neurotherapeutics. 2016 Oct;13(4):748-761. doi: 10.1007/s13311-016-0465-z. Neurotherapeutics. 2016. PMID: 27525700 Free PMC article. Review.
Cited by
-
Ischemic preconditioning provides long-lasting neuroprotection against ischemic stroke: The role of Nrf2.Exp Neurol. 2020 Mar;325:113142. doi: 10.1016/j.expneurol.2019.113142. Epub 2019 Dec 5. Exp Neurol. 2020. PMID: 31812555 Free PMC article.
-
Microglia activation mediated by toll-like receptor-4 impairs brain white matter tracts in rats.J Biomed Res. 2018 Mar 26;32(2):136-144. doi: 10.7555/JBR.32.20170033. J Biomed Res. 2018. PMID: 29358565 Free PMC article.
-
Memory-Like Inflammatory Responses of Microglia to Rising Doses of LPS: Key Role of PI3Kγ.Front Immunol. 2019 Nov 8;10:2492. doi: 10.3389/fimmu.2019.02492. eCollection 2019. Front Immunol. 2019. PMID: 31781091 Free PMC article.
-
A method for reducing animal use whilst maintaining statistical power in electrophysiological recordings from rodent nerves.Heliyon. 2020 Jun 6;6(6):e04143. doi: 10.1016/j.heliyon.2020.e04143. eCollection 2020 Jun. Heliyon. 2020. PMID: 32529085 Free PMC article.
-
NogoA Neutralization Promotes Axonal Restoration After White Matter Injury In Subcortical Stroke.Sci Rep. 2017 Aug 25;7(1):9431. doi: 10.1038/s41598-017-09705-0. Sci Rep. 2017. PMID: 28842591 Free PMC article.
References
-
- Bahjat FR, Williams-Karnesky RL, Kohama SG, West GA, Doyle KP, Spector MD, Hobbs TR, Stenzel-Poore MP. Proof of concept: pharmacological preconditioning with a Toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab. 2011;31:1229–1242. doi: 10.1038/jcbfm.2011.6. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
