

Nuclear Factor of Activated T Cells-dependent Down-regulation of the Transcription Factor Glioma-associated Protein 1 (GLI1) Underlies the Growth Inhibitory Properties of Arachidonic Acid
- PMID: 26601952
- PMCID: PMC4722469
- DOI: 10.1074/jbc.M115.691972
Nuclear Factor of Activated T Cells-dependent Down-regulation of the Transcription Factor Glioma-associated Protein 1 (GLI1) Underlies the Growth Inhibitory Properties of Arachidonic Acid
Abstract
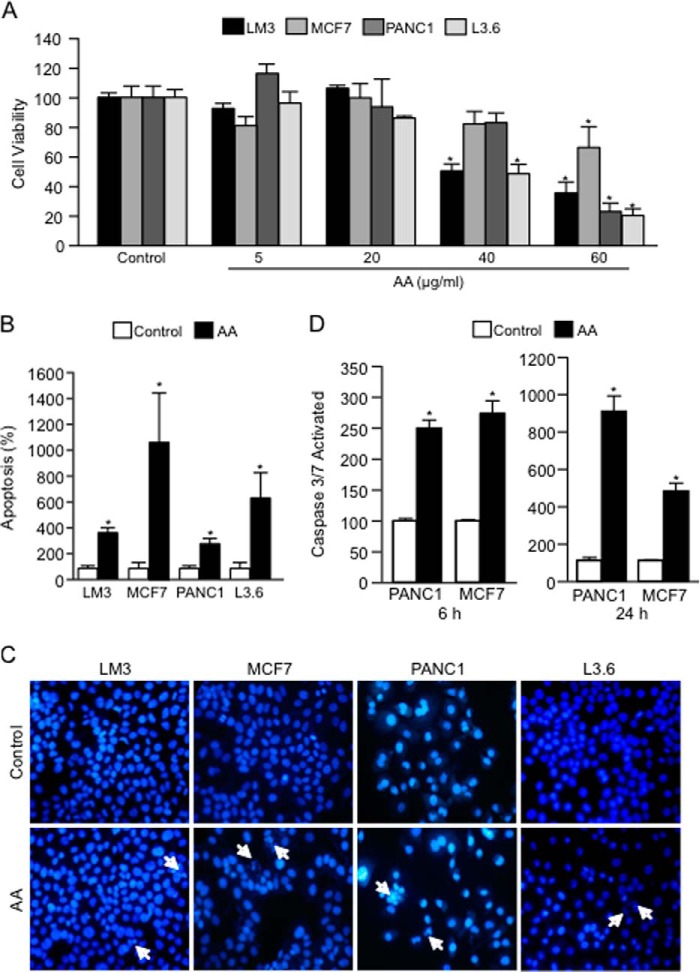
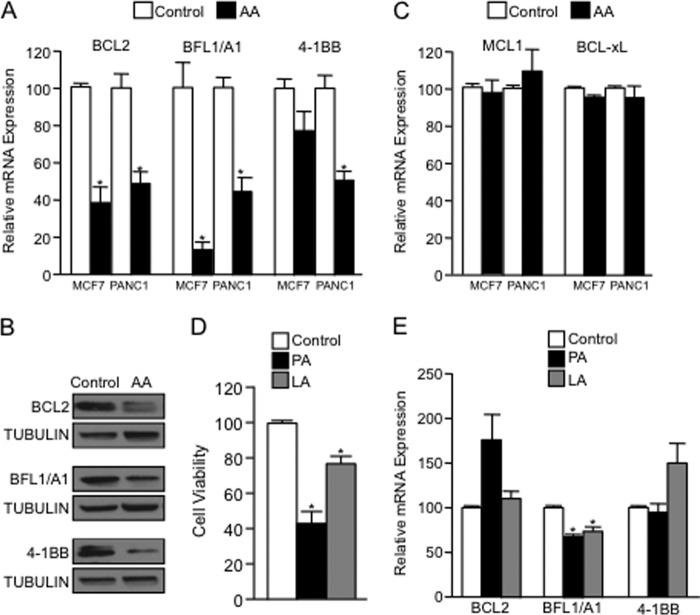
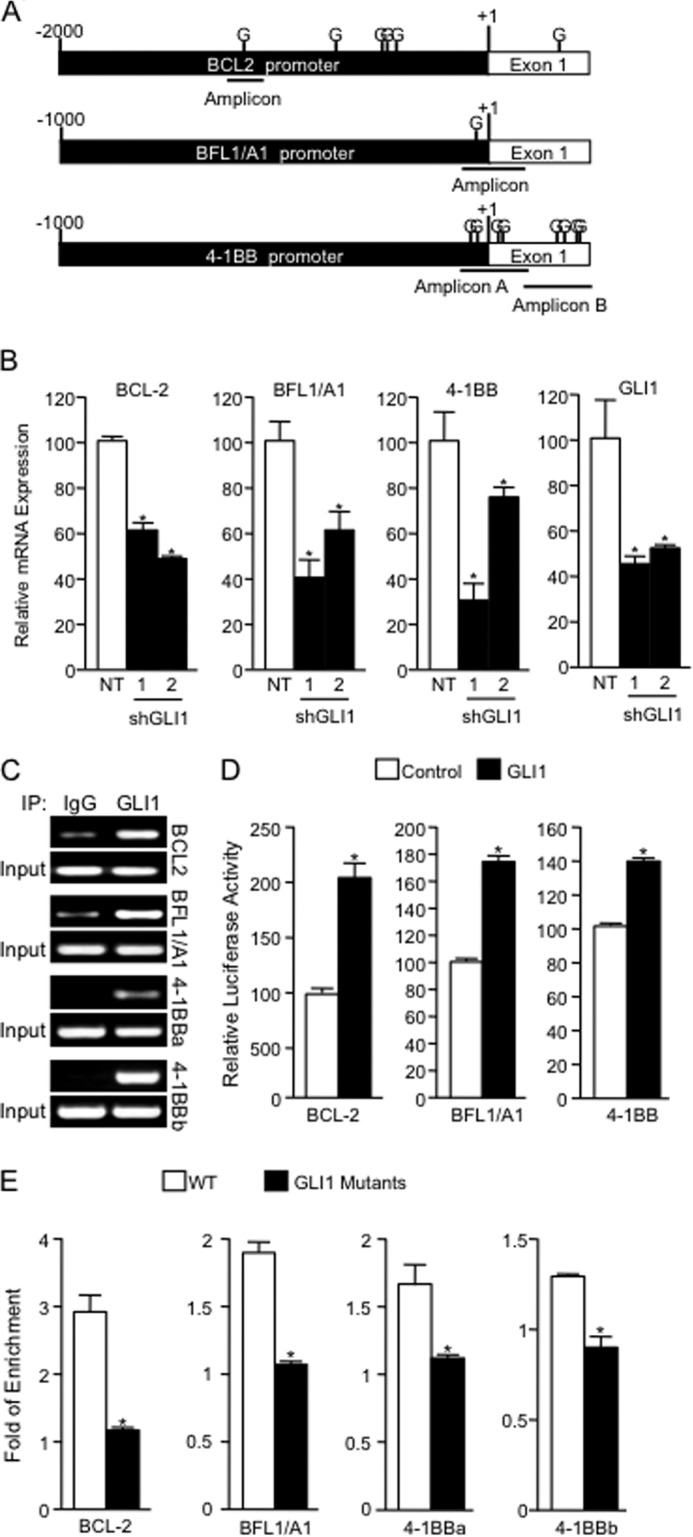
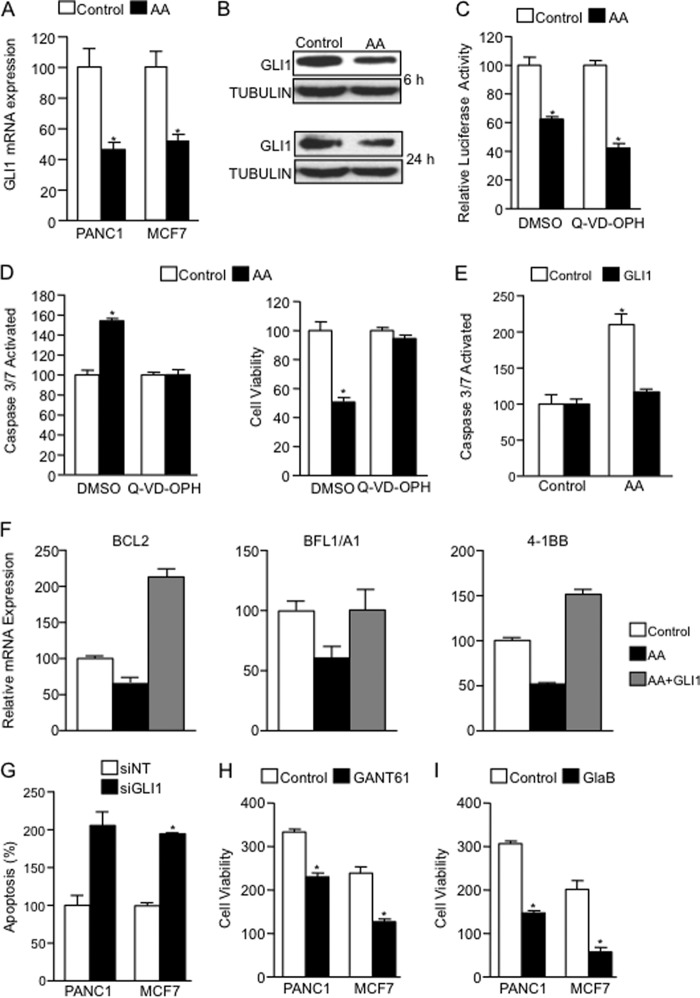
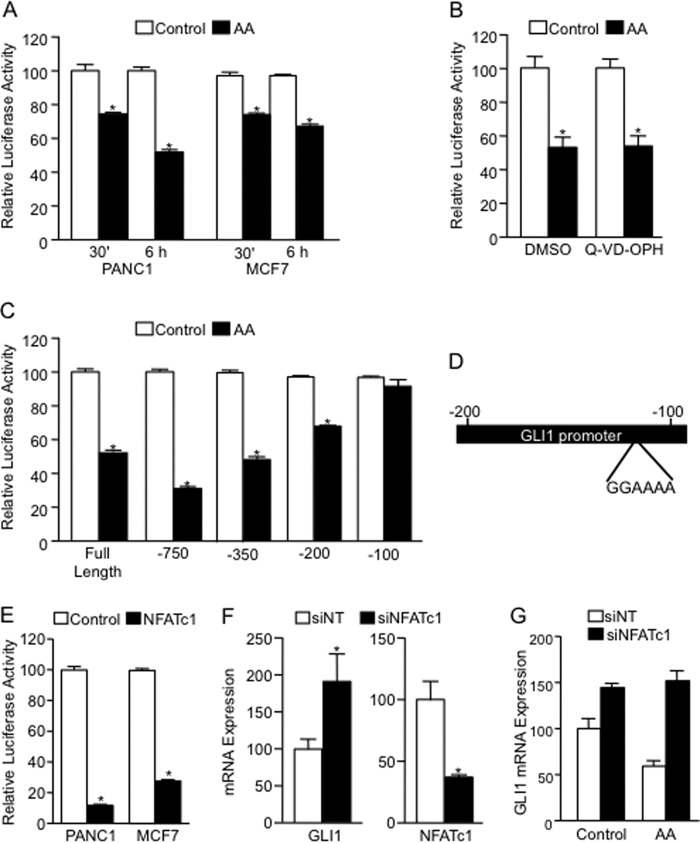
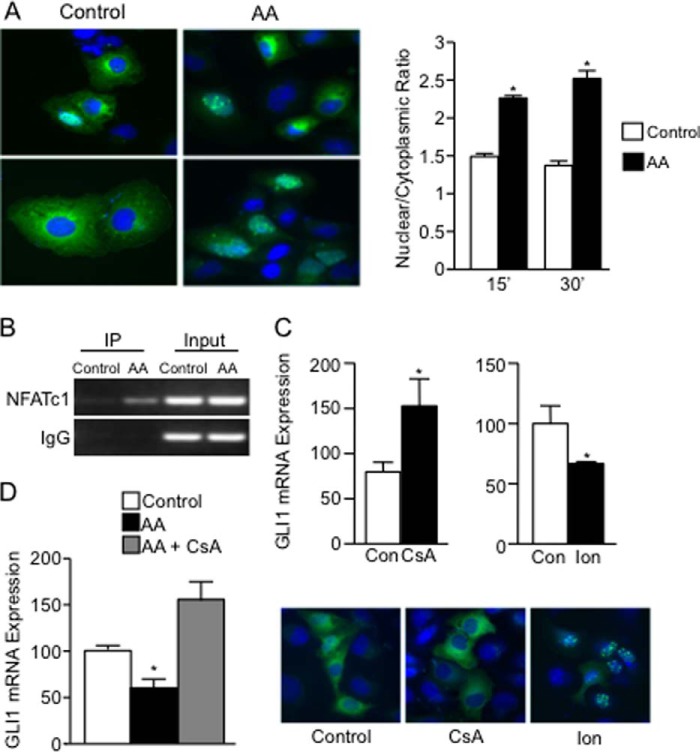
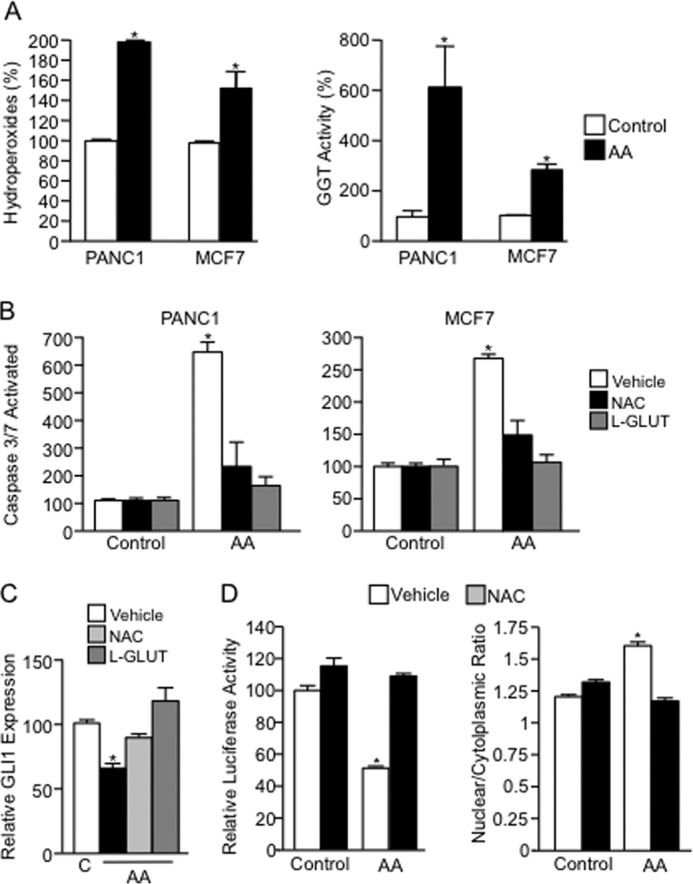
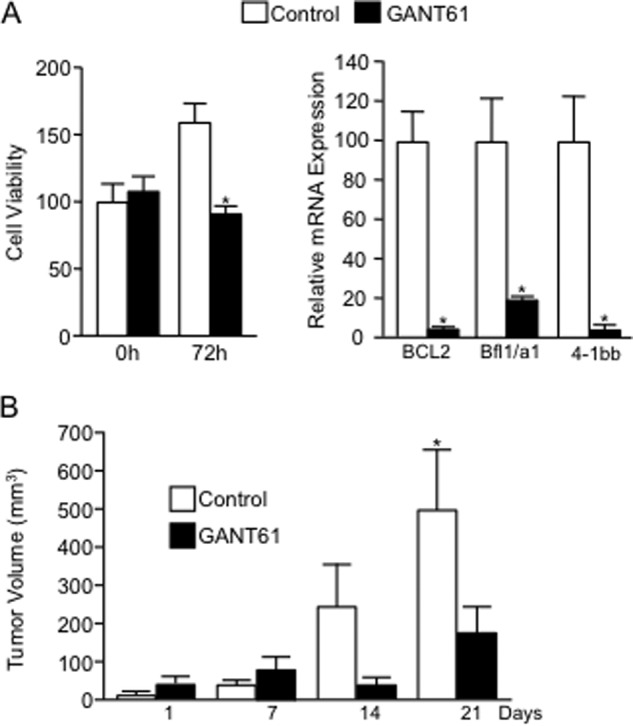
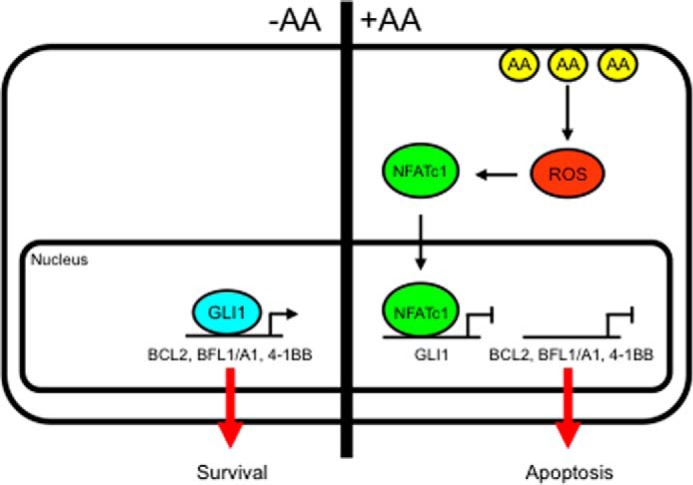
Numerous reports have demonstrated a tumor inhibitory effect of polyunsaturated fatty acids (PUFAs). However, the molecular mechanisms modulating this phenomenon are in part poorly understood. Here, we provide evidence of a novel antitumoral mechanism of the PUFA arachidonic acid (AA). In vivo and in vitro experiments showed that AA treatment decreased tumor growth and metastasis and increased apoptosis. Molecular analysis of this effect showed significantly reduced expression of a subset of antiapoptotic proteins, including BCL2, BFL1/A1, and 4-1BB, in AA-treated cells. We demonstrated that down-regulation of the transcription factor glioma-associated protein 1 (GLI1) in AA-treated cells is the underlying mechanism controlling BCL2, BFL1/A1, and 4-1BB expression. Using luciferase reporters, chromatin immunoprecipitation, and expression studies, we found that GLI1 binds to the promoter of these antiapoptotic molecules and regulates their expression and promoter activity. We provide evidence that AA-induced apoptosis and down-regulation of antiapoptotic genes can be inhibited by overexpressing GLI1 in AA-sensitive cells. Conversely, inhibition of GLI1 mimics AA treatments, leading to decreased tumor growth, cell viability, and expression of antiapoptotic molecules. Further characterization showed that AA represses GLI1 expression by stimulating nuclear translocation of NFATc1, which then binds the GLI1 promoter and represses its transcription. AA was shown to increase reactive oxygen species. Treatment with antioxidants impaired the AA-induced apoptosis and down-regulation of GLI1 and NFATc1 activation, indicating that NFATc1 activation and GLI1 repression require the generation of reactive oxygen species. Collectively, these results define a novel mechanism underlying AA antitumoral functions that may serve as a foundation for future PUFA-based therapeutic approaches.
Keywords: GLI1; arachidonic acid (AA) (ARA); cancer; cell death; polyunsaturated fatty acid (PUFA); transcription factor.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures

Similar articles
-
Forkhead Box M1 Is Essential for Nuclear Localization of Glioma-associated Oncogene Homolog 1 in Glioblastoma Multiforme Cells by Promoting Importin-7 Expression.J Biol Chem. 2015 Jul 24;290(30):18662-70. doi: 10.1074/jbc.M115.662882. Epub 2015 Jun 17. J Biol Chem. 2015. PMID: 26085085 Free PMC article.
-
Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma.Cell Death Dis. 2015 Apr 9;6(4):e1712. doi: 10.1038/cddis.2015.76. Cell Death Dis. 2015. PMID: 25855960 Free PMC article.
-
Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma.Mol Cancer Res. 2013 Jun;11(6):604-15. doi: 10.1158/1541-7786.MCR-12-0441. Epub 2013 Mar 22. Mol Cancer Res. 2013. PMID: 23525267
-
Aberrant activation of hedgehog signaling promotes cell proliferation via the transcriptional activation of forkhead Box M1 in colorectal cancer cells.J Exp Clin Cancer Res. 2017 Feb 2;36(1):23. doi: 10.1186/s13046-017-0491-7. J Exp Clin Cancer Res. 2017. PMID: 28148279 Free PMC article.
-
Transcription factor-mediated epigenetic regulation of cell growth and phenotype for biological control and cancer.Adv Enzyme Regul. 2010;50(1):160-7. doi: 10.1016/j.advenzreg.2009.10.026. Epub 2009 Nov 4. Adv Enzyme Regul. 2010. PMID: 19896493 Free PMC article. Review. No abstract available.
Cited by
-
Arginylated Calreticulin Increases Apoptotic Response Induced by Bortezomib in Glioma Cells.Mol Neurobiol. 2019 Mar;56(3):1653-1664. doi: 10.1007/s12035-018-1182-x. Epub 2018 Jun 18. Mol Neurobiol. 2019. PMID: 29916141
-
Polyunsaturated Fatty Acid-Enriched Lipid Fingerprint of Glioblastoma Proliferative Regions Is Differentially Regulated According to Glioblastoma Molecular Subtype.Int J Mol Sci. 2022 Mar 9;23(6):2949. doi: 10.3390/ijms23062949. Int J Mol Sci. 2022. PMID: 35328369 Free PMC article.
-
Quinolines and Oxazino-quinoline Derivatives as Small Molecule GLI1 Inhibitors Identified by Virtual Screening.ACS Med Chem Lett. 2022 Jul 22;13(8):1329-1336. doi: 10.1021/acsmedchemlett.2c00249. eCollection 2022 Aug 11. ACS Med Chem Lett. 2022. PMID: 35978701 Free PMC article.
-
GLI1: A Therapeutic Target for Cancer.Front Oncol. 2021 May 25;11:673154. doi: 10.3389/fonc.2021.673154. eCollection 2021. Front Oncol. 2021. PMID: 34113570 Free PMC article. Review.
-
RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis.Nat Commun. 2021 May 18;12(1):2901. doi: 10.1038/s41467-021-23186-w. Nat Commun. 2021. PMID: 34006870 Free PMC article.
References
-
- Dahm C. C., Gorst-Rasmussen A., Crowe F. L., Roswall N., Tjønneland A., Drogan D., Boeing H., Teucher B., Kaaks R., Adarakis G., Zylis D., Trichopoulou A., Fedirko V., Chajes V., Jenab M., Palli D., Pala V., Tumino R., Ricceri F., van Kranen H., Bueno-de-Mesquita H. B., Quirós J. R., Sánchez M. J., Luján-Barroso L., Larrañaga N., Chirlaque M. D., Ardanaz E., Johansson M., Stattin P., Khaw K. T., Wareham N., Wark P. A., Norat T., Riboli E., Key T. J., and Overvad K. (2012) Fatty acid patterns and risk of prostate cancer in a case-control study nested within the European Prospective Investigation into Cancer and Nutrition. Am. J. Clin. Nutr. 96, 1354–1361 - PubMed
-
- Eynard A. R., and Navarro A. (2013) Crosstalk among dietary polyunsaturated fatty acids, urolithiasis, chronic inflammation, and urinary tract tumor risk. Nutrition 29, 930–938 - PubMed
-
- Tapiero H., Ba G. N., Couvreur P., and Tew K. D. (2002) Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed. Pharmacother. 56, 215–222 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
