

Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci
- PMID: 26402605
- PMCID: PMC4624267
- DOI: 10.1016/j.neuron.2015.09.016
Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci
Abstract
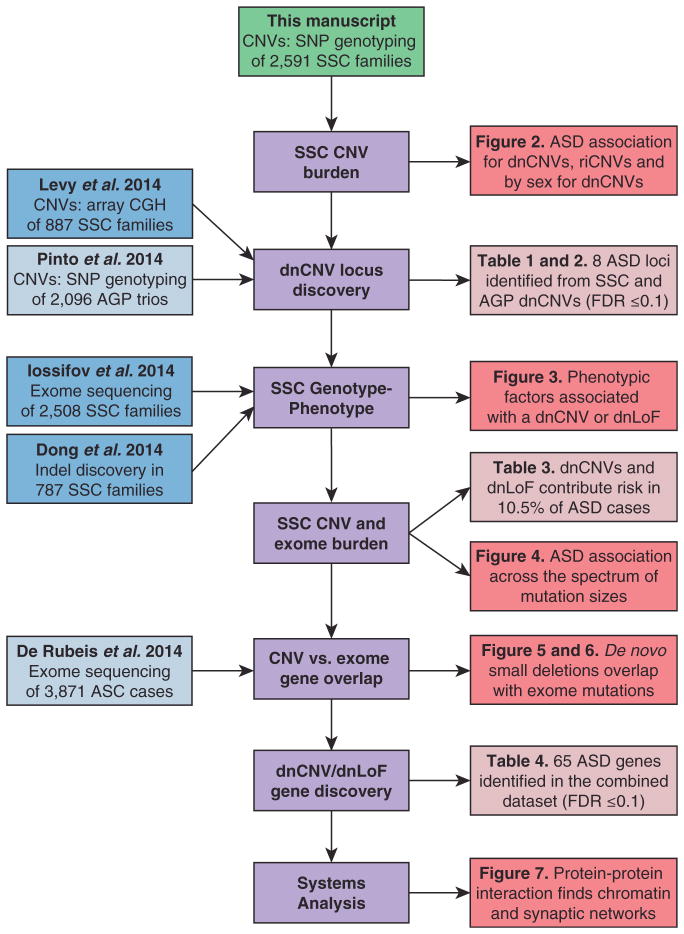
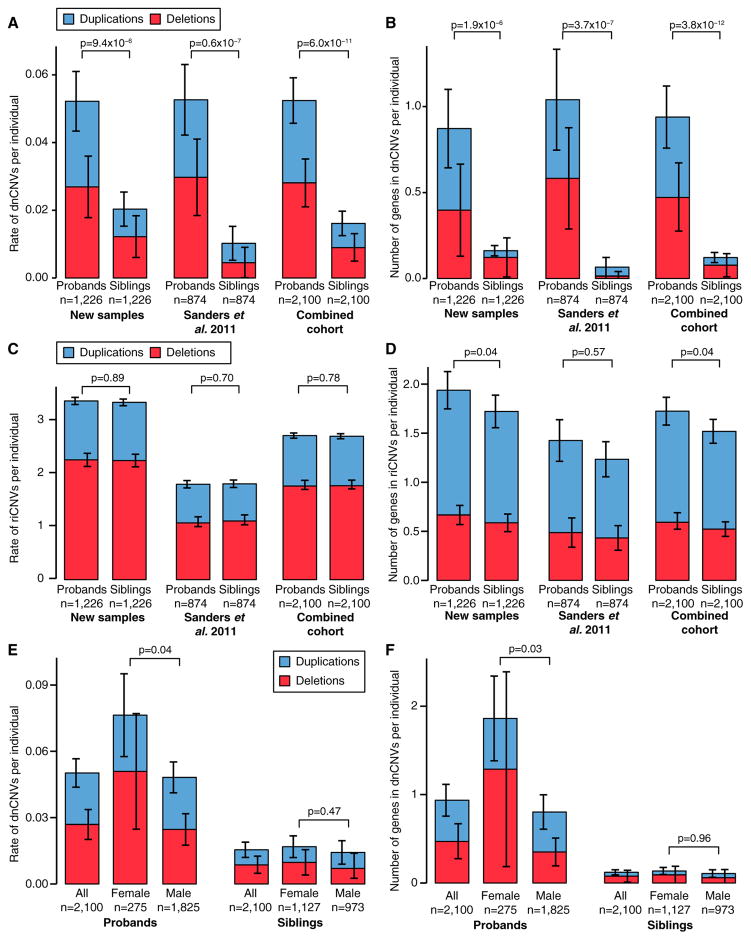
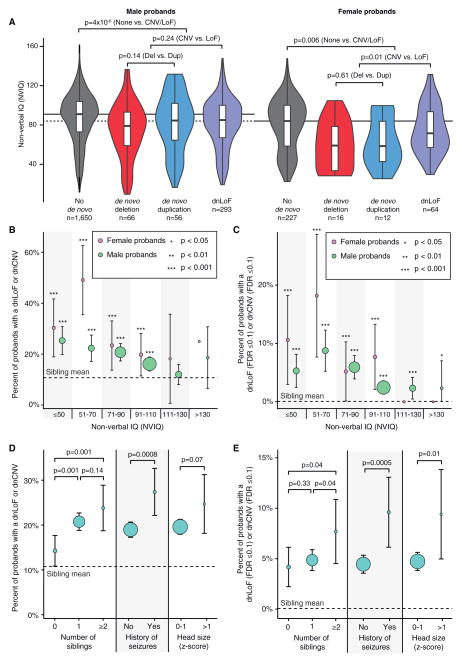
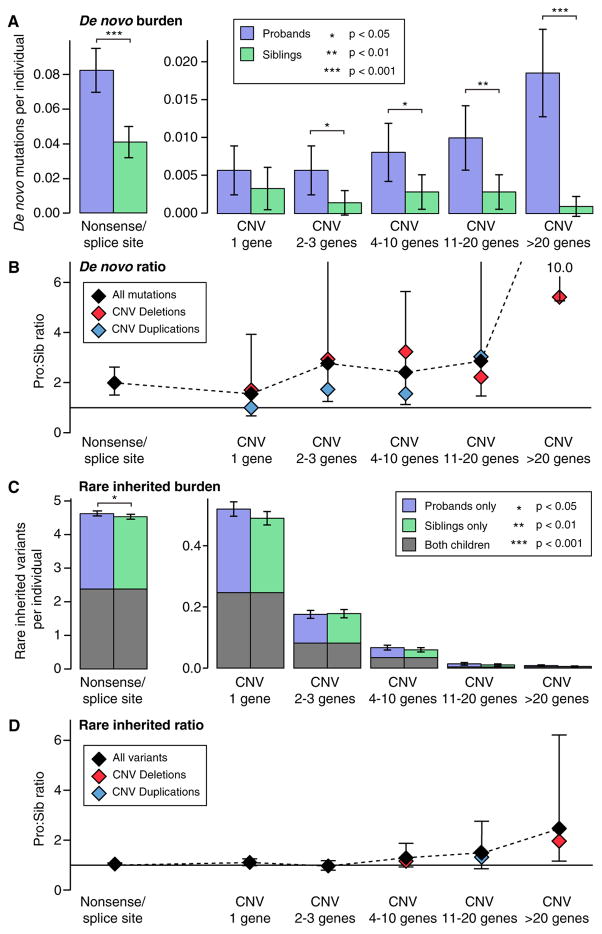
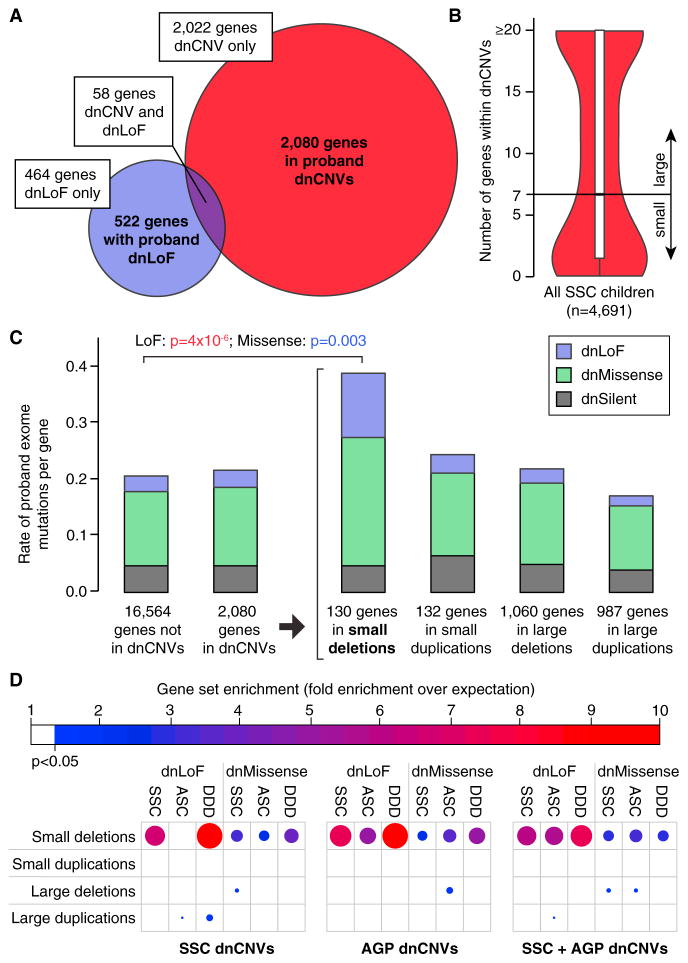
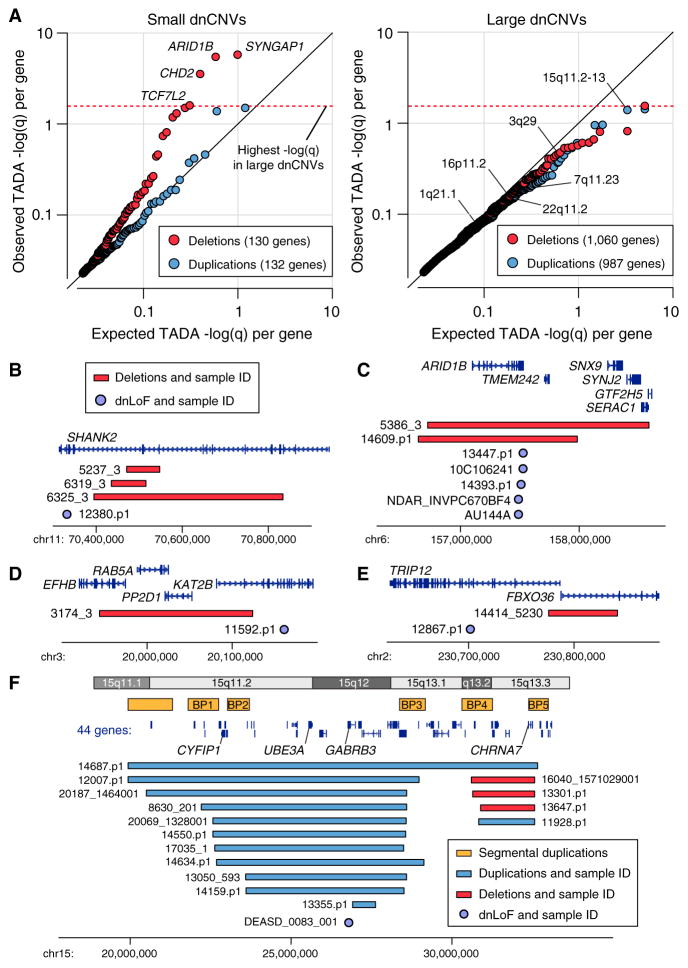
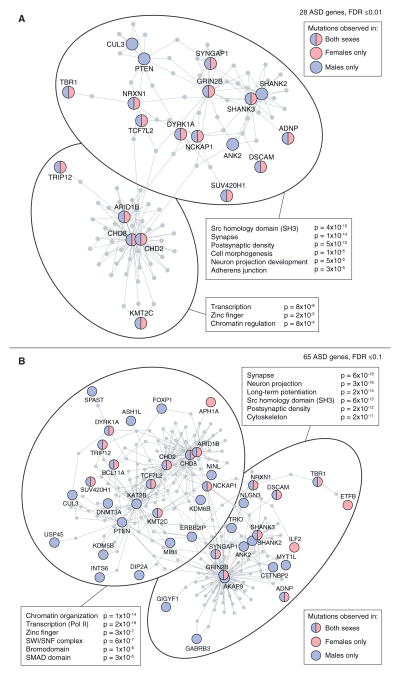
Analysis of de novo CNVs (dnCNVs) from the full Simons Simplex Collection (SSC) (N = 2,591 families) replicates prior findings of strong association with autism spectrum disorders (ASDs) and confirms six risk loci (1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2-13, and 22q11.2). The addition of published CNV data from the Autism Genome Project (AGP) and exome sequencing data from the SSC and the Autism Sequencing Consortium (ASC) shows that genes within small de novo deletions, but not within large dnCNVs, significantly overlap the high-effect risk genes identified by sequencing. Alternatively, large dnCNVs are found likely to contain multiple modest-effect risk genes. Overall, we find strong evidence that de novo mutations are associated with ASD apart from the risk for intellectual disability. Extending the transmission and de novo association test (TADA) to include small de novo deletions reveals 71 ASD risk loci, including 6 CNV regions (noted above) and 65 risk genes (FDR ≤ 0.1).
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Meta-Analyses Support Previous and Novel Autism Candidate Genes: Outcomes of an Unexplored Brazilian Cohort.Autism Res. 2020 Feb;13(2):199-206. doi: 10.1002/aur.2238. Epub 2019 Nov 6. Autism Res. 2020. PMID: 31696658
-
Integrated Functional Analysis Implicates Syndromic and Rare Copy Number Variation Genes as Prominent Molecular Players in Pathogenesis of Autism Spectrum Disorders.Neuroscience. 2020 Jul 1;438:25-40. doi: 10.1016/j.neuroscience.2020.04.051. Epub 2020 May 12. Neuroscience. 2020. PMID: 32407977
-
Prevalence and phenotypic impact of rare potentially damaging variants in autism spectrum disorder.Mol Autism. 2021 Oct 6;12(1):65. doi: 10.1186/s13229-021-00465-3. Mol Autism. 2021. PMID: 34615535 Free PMC article.
-
Genetic architecture of autism spectrum disorder: Lessons from large-scale genomic studies.Neurosci Biobehav Rev. 2021 Sep;128:244-257. doi: 10.1016/j.neubiorev.2021.06.028. Epub 2021 Jun 21. Neurosci Biobehav Rev. 2021. PMID: 34166716 Review.
-
The genetics of autism.Pediatrics. 2004 May;113(5):e472-86. doi: 10.1542/peds.113.5.e472. Pediatrics. 2004. PMID: 15121991 Review.
Cited by
-
Genetics of autism spectrum disorders and future direction.J Hum Genet. 2023 Mar;68(3):193-197. doi: 10.1038/s10038-022-01076-3. Epub 2022 Aug 30. J Hum Genet. 2023. PMID: 36038624 Review.
-
Thalamocortical organoids enable in vitro modeling of 22q11.2 microdeletion associated with neuropsychiatric disorders.Cell Stem Cell. 2024 Mar 7;31(3):421-432.e8. doi: 10.1016/j.stem.2024.01.010. Epub 2024 Feb 20. Cell Stem Cell. 2024. PMID: 38382530
-
Covariance-based sample selection for heterogeneous data: Applications to gene expression and autism risk gene detection.J Am Stat Assoc. 2021;116(533):54-67. doi: 10.1080/01621459.2020.1738234. Epub 2020 Apr 13. J Am Stat Assoc. 2021. PMID: 33731968 Free PMC article.
-
Genome-wide detection of tandem DNA repeats that are expanded in autism.Nature. 2020 Oct;586(7827):80-86. doi: 10.1038/s41586-020-2579-z. Epub 2020 Jul 27. Nature. 2020. PMID: 32717741 Free PMC article.
-
Delving into the Complexity of Valproate-Induced Autism Spectrum Disorder: The Use of Zebrafish Models.Cells. 2024 Aug 14;13(16):1349. doi: 10.3390/cells13161349. Cells. 2024. PMID: 39195239 Free PMC article.
References
-
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. - PubMed
Publication types
MeSH terms
Grants and funding
- MH19961-14/MH/NIMH NIH HHS/United States
- R01 MH074090/MH/NIMH NIH HHS/United States
- MH100233/MH/NIMH NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- R01 MH097849/MH/NIMH NIH HHS/United States
- MH100209/MH/NIMH NIH HHS/United States
- U01 MH100229/MH/NIMH NIH HHS/United States
- MH074090/MH/NIMH NIH HHS/United States
- R01 DC009410/DC/NIDCD NIH HHS/United States
- T32 MH019961/MH/NIMH NIH HHS/United States
- U01 MH100209/MH/NIMH NIH HHS/United States
- U01 MH100239/MH/NIMH NIH HHS/United States
- DC009410/DC/NIDCD NIH HHS/United States
- MH100239/MH/NIMH NIH HHS/United States
- MH100229/MH/NIMH NIH HHS/United States
- R01 MH057881/MH/NIMH NIH HHS/United States
- S10 OD018522/OD/NIH HHS/United States
- P30 DC005188/DC/NIDCD NIH HHS/United States
- Canadian Institutes of Health Research/Canada
- MH071584-07/MH/NIMH NIH HHS/United States
- R25 MH071584/MH/NIMH NIH HHS/United States
- T32 MH014276/MH/NIMH NIH HHS/United States
- Howard Hughes Medical Institute/United States
- MH057881/MH/NIMH NIH HHS/United States
- U01 MH100233/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
