

HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation
- PMID: 26387755
- PMCID: PMC4636961
- DOI: 10.1016/j.stem.2015.08.004
HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation
Abstract
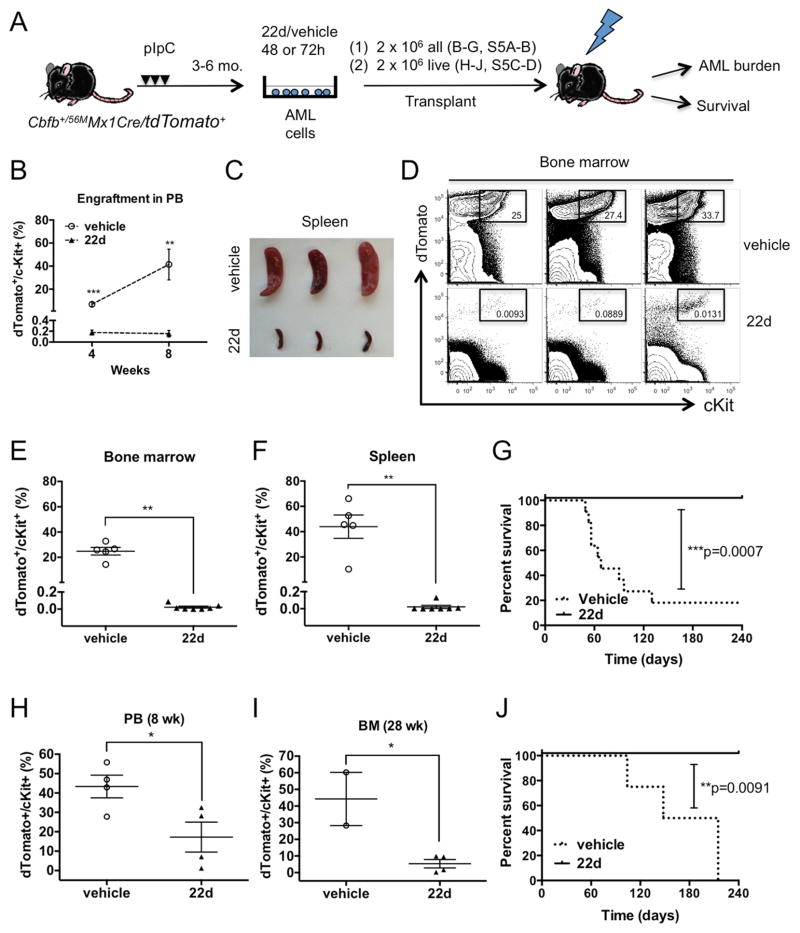
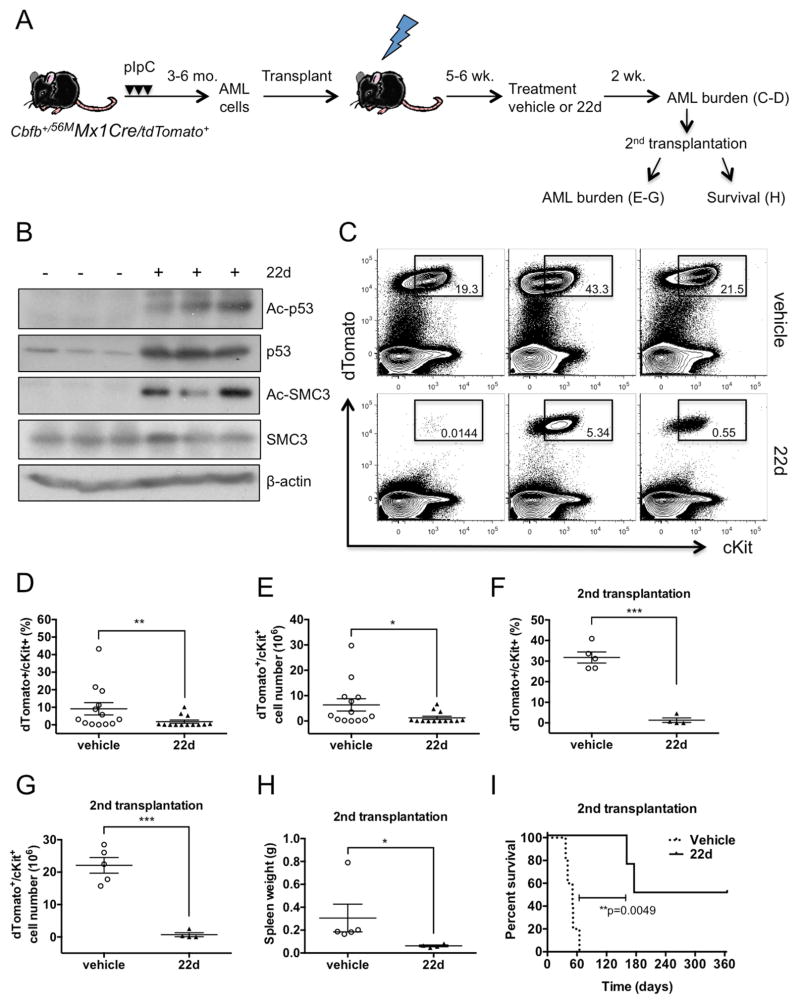
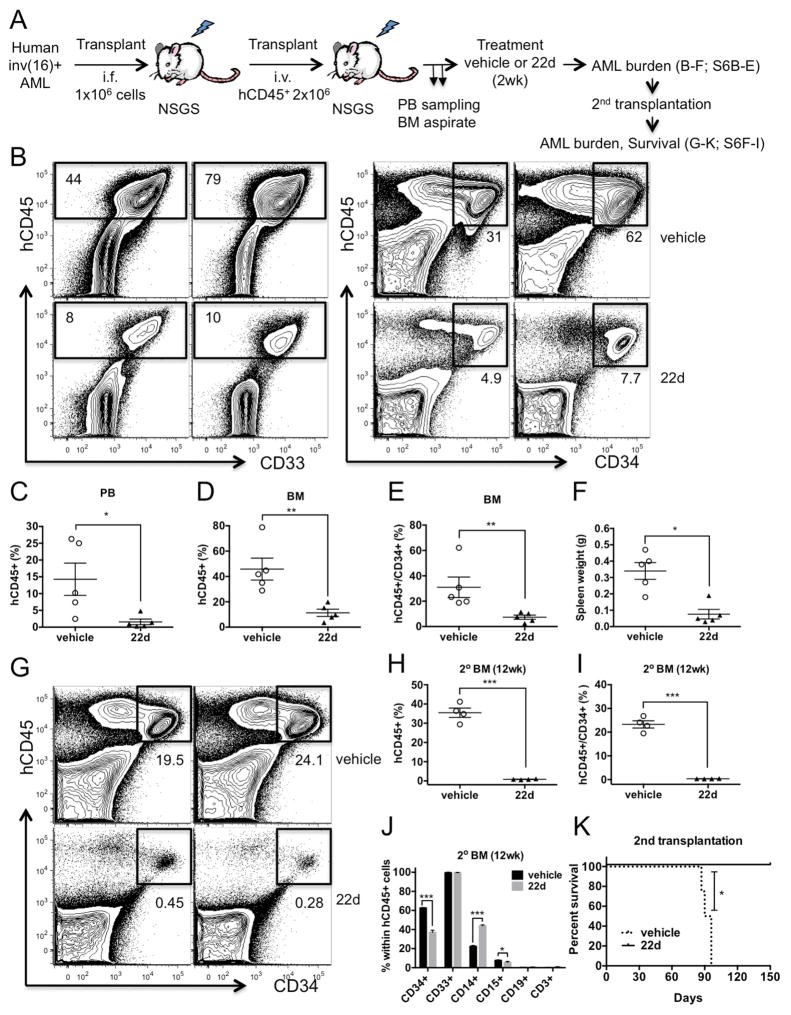
Acute myeloid leukemia (AML) is driven and sustained by leukemia stem cells (LSCs) with unlimited self-renewal capacity and resistance to chemotherapy. Mutation in the TP53 tumor suppressor is relatively rare in de novo AML; however, p53 can be regulated through post-translational mechanisms. Here, we show that p53 activity is inhibited in inv(16)(+) AML LSCs via interactions with the CBFβ-SMMHC (CM) fusion protein and histone deacetylase 8 (HDAC8). HDAC8 aberrantly deacetylates p53 and promotes LSC transformation and maintenance. HDAC8 deficiency or inhibition using HDAC8-selective inhibitors (HDAC8i) effectively restores p53 acetylation and activity. Importantly, HDAC8 inhibition induces apoptosis in inv(16)(+) AML CD34(+) cells, while sparing the normal hematopoietic stem cells. Furthermore, in vivo HDAC8i administration profoundly diminishes AML propagation and abrogates leukemia-initiating capacity of both murine and patient-derived LSCs. This study elucidates an HDAC8-mediated p53-inactivating mechanism promoting LSC activity and highlights HDAC8 inhibition as a promising approach to selectively target inv(16)(+) LSCs.
Copyright © 2015 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declared that no conflicts of interest exist.
Figures
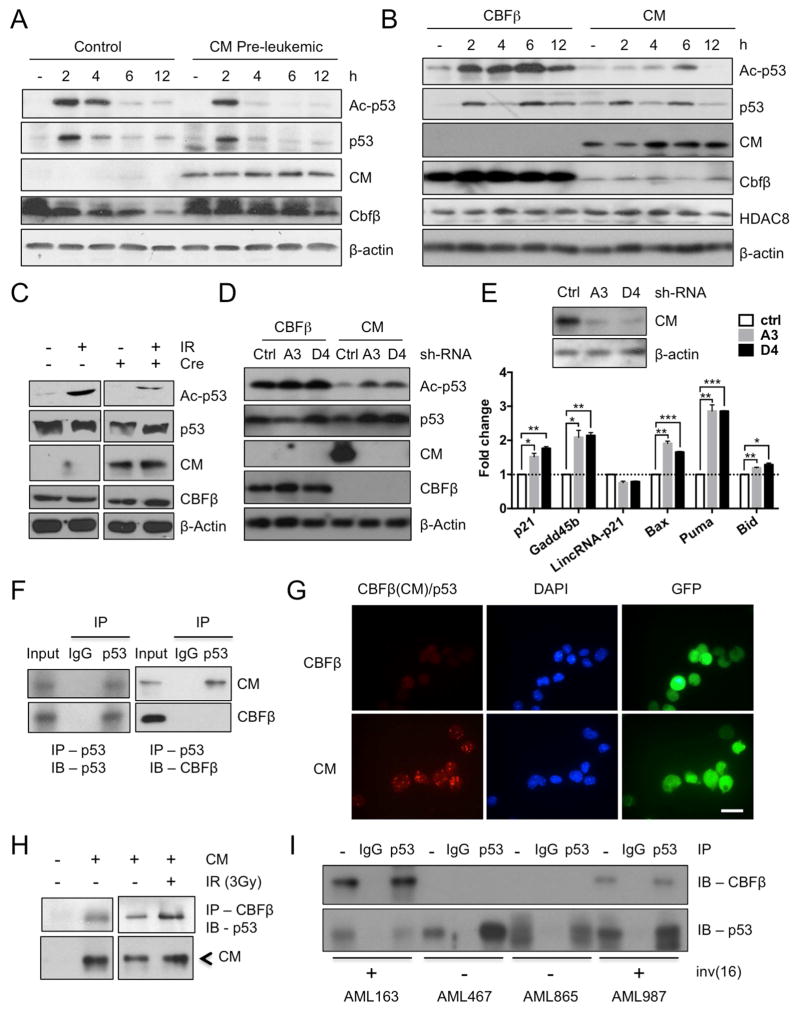
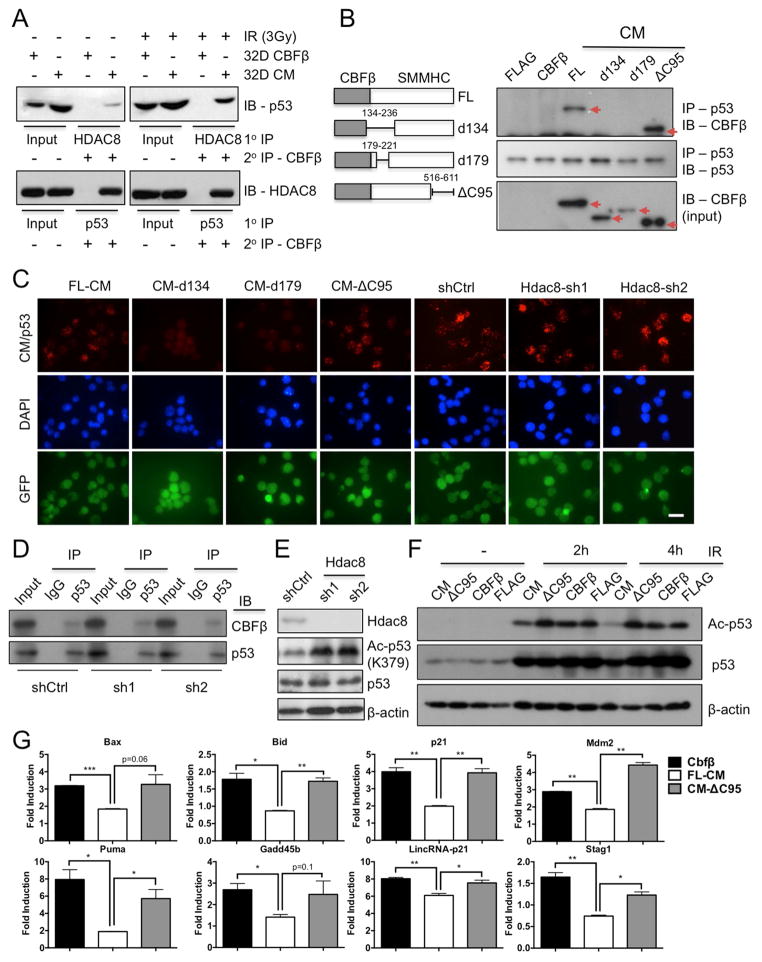
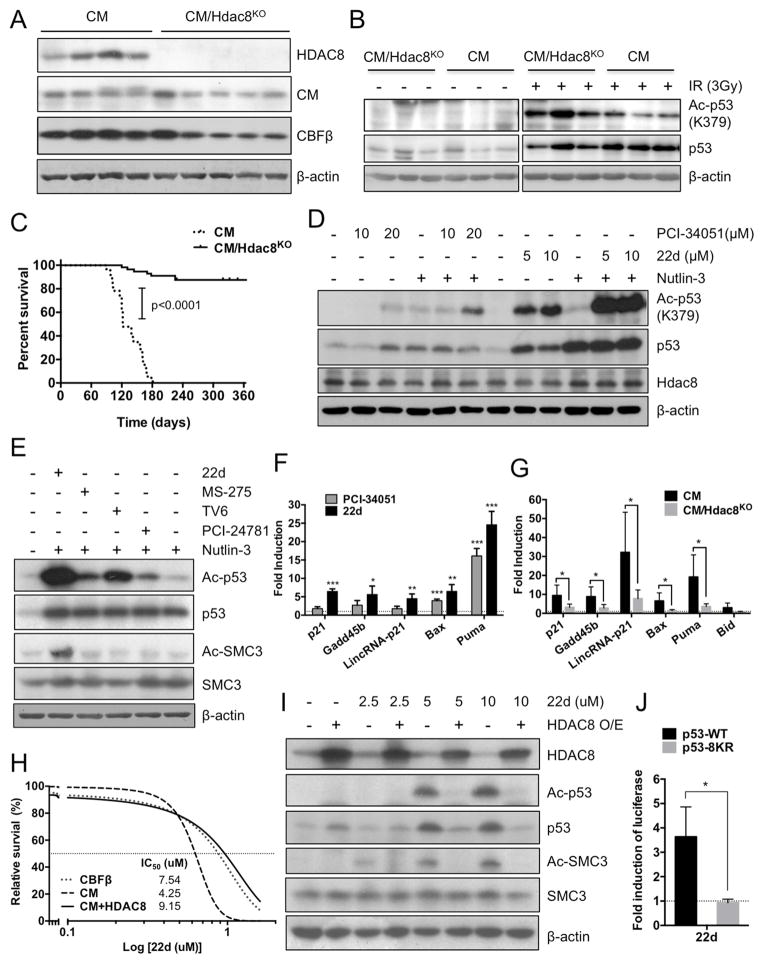
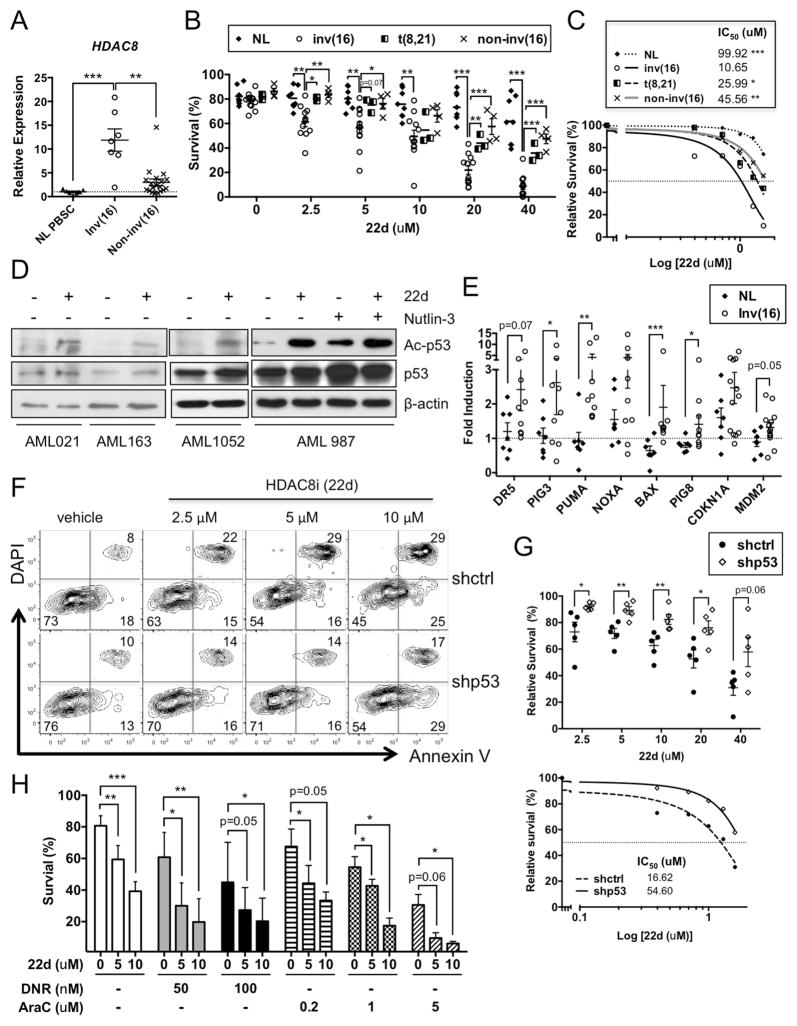
Similar articles
-
FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia.Blood. 2020 Apr 23;135(17):1472-1483. doi: 10.1182/blood.2019003538. Blood. 2020. PMID: 32315388
-
Targeting miR-126 in inv(16) acute myeloid leukemia inhibits leukemia development and leukemia stem cell maintenance.Nat Commun. 2021 Oct 22;12(1):6154. doi: 10.1038/s41467-021-26420-7. Nat Commun. 2021. PMID: 34686664 Free PMC article.
-
Regain control of p53: Targeting leukemia stem cells by isoform-specific HDAC inhibition.Exp Hematol. 2016 May;44(5):315-21. doi: 10.1016/j.exphem.2016.02.007. Epub 2016 Feb 26. Exp Hematol. 2016. PMID: 26923266 Free PMC article. Review.
-
Targeting binding partners of the CBFβ-SMMHC fusion protein for the treatment of inversion 16 acute myeloid leukemia.Oncotarget. 2016 Oct 4;7(40):66255-66266. doi: 10.18632/oncotarget.11357. Oncotarget. 2016. PMID: 27542261 Free PMC article. Review.
-
Are inhibitors of histone deacetylase 8 (HDAC8) effective in hematological cancers especially acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL)?Eur J Med Chem. 2023 Oct 5;258:115594. doi: 10.1016/j.ejmech.2023.115594. Epub 2023 Jun 25. Eur J Med Chem. 2023. PMID: 37429084 Review.
Cited by
-
ZNF521 promotes acute myeloid leukemogenesis by suppressing the expression and acetylation of SMC3.Heliyon. 2024 Sep 5;10(18):e37528. doi: 10.1016/j.heliyon.2024.e37528. eCollection 2024 Sep 30. Heliyon. 2024. PMID: 39309877 Free PMC article.
-
Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity.Front Genet. 2019 Mar 1;10:133. doi: 10.3389/fgene.2019.00133. eCollection 2019. Front Genet. 2019. PMID: 30881380 Free PMC article. Review.
-
Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy.Blood. 2017 Aug 10;130(6):699-712. doi: 10.1182/blood-2017-02-763086. Epub 2017 Jun 12. Blood. 2017. PMID: 28607134 Free PMC article. Review.
-
Proteogenomic analysis reveals cytoplasmic sequestration of RUNX1 by the acute myeloid leukemia-initiating CBFB::MYH11 oncofusion protein.J Clin Invest. 2023 Dec 7;134(4):e176311. doi: 10.1172/JCI176311. J Clin Invest. 2023. PMID: 38061017 Free PMC article.
-
HDAC1 Is a Required Cofactor of CBFβ-SMMHC and a Potential Therapeutic Target in Inversion 16 Acute Myeloid Leukemia.Mol Cancer Res. 2019 Jun;17(6):1241-1252. doi: 10.1158/1541-7786.MCR-18-0922. Epub 2019 Feb 27. Mol Cancer Res. 2019. PMID: 30814129 Free PMC article.
References
-
- Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–034. - PubMed
-
- Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, Lotem J, Tanay A, Groner Y. Addiction of t(8;21) and inv(16) Acute Myeloid Leukemia to Native RUNX1. Cell Rep. 2013;4:1131–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
