

Necroinflammation in Kidney Disease
- PMID: 26334031
- PMCID: PMC4696588
- DOI: 10.1681/ASN.2015040405
Necroinflammation in Kidney Disease
Abstract
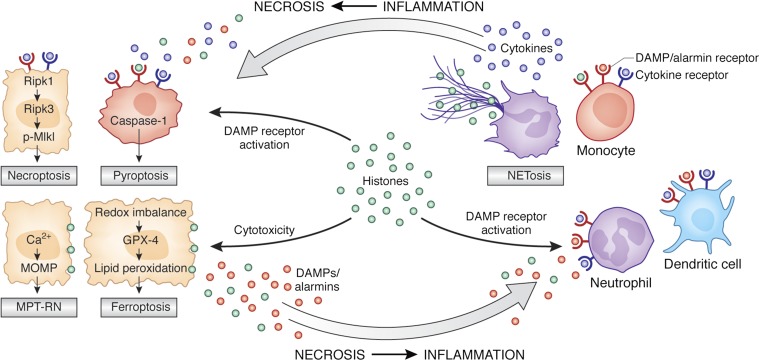
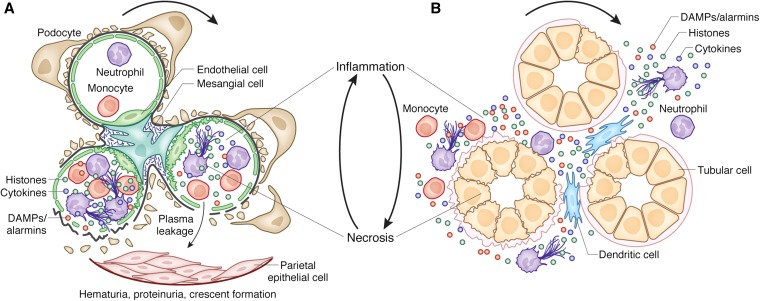
The bidirectional causality between kidney injury and inflammation remains an area of unexpected discoveries. The last decade unraveled the molecular mechanisms of sterile inflammation, which established danger signaling via pattern recognition receptors as a new concept of kidney injury-related inflammation. In contrast, renal cell necrosis remained considered a passive process executed either by the complement-related membrane attack complex, exotoxins, or cytotoxic T cells. Accumulating data now suggest that renal cell necrosis is a genetically determined and regulated process involving specific outside-in signaling pathways. These findings support a unifying theory in which kidney injury and inflammation are reciprocally enhanced in an autoamplification loop, referred to here as necroinflammation. This integrated concept is of potential clinical importance because it offers numerous innovative molecular targets for limiting kidney injury by blocking cell death, inflammation, or both. Here, the contribution of necroinflammation to AKI is discussed in thrombotic microangiopathies, necrotizing and crescentic GN, acute tubular necrosis, and infective pyelonephritis or sepsis. Potential new avenues are further discussed for abrogating necroinflammation-related kidney injury, and questions and strategies are listed for further exploration in this evolving field.
Keywords: Immunology and pathology; acute renal failure; kidney failure.
Copyright © 2016 by the American Society of Nephrology.
Figures
Similar articles
-
Cell Death Pathways Drive Necroinflammation during Acute Kidney Injury.Nephron. 2018;140(2):144-147. doi: 10.1159/000490807. Epub 2018 Jun 29. Nephron. 2018. PMID: 29961062 Review.
-
How Kidney Cell Death Induces Renal Necroinflammation.Semin Nephrol. 2016 May;36(3):162-73. doi: 10.1016/j.semnephrol.2016.03.004. Semin Nephrol. 2016. PMID: 27339382 Review.
-
Extracellular traps in kidney disease.Kidney Int. 2018 Dec;94(6):1087-1098. doi: 10.1016/j.kint.2018.08.035. Kidney Int. 2018. PMID: 30466565 Review.
-
SAP130 released by damaged tubule drives necroinflammation via miRNA-219c/Mincle signaling in acute kidney injury.Cell Death Dis. 2021 Sep 23;12(10):866. doi: 10.1038/s41419-021-04131-7. Cell Death Dis. 2021. PMID: 34556635 Free PMC article.
-
Different Patterns of Kidney Fibrosis Are Indicative of Injury to Distinct Renal Compartments.Cells. 2021 Aug 6;10(8):2014. doi: 10.3390/cells10082014. Cells. 2021. PMID: 34440782 Free PMC article.
Cited by
-
Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases.Medicina (Kaunas). 2019 Aug 2;55(8):431. doi: 10.3390/medicina55080431. Medicina (Kaunas). 2019. PMID: 31382486 Free PMC article. Review.
-
Necroinflammation emerges as a key regulator of hematopoiesis in health and disease.Cell Death Differ. 2019 Jan;26(1):53-67. doi: 10.1038/s41418-018-0194-4. Epub 2018 Sep 21. Cell Death Differ. 2019. PMID: 30242210 Free PMC article. Review.
-
Postoperative cellular stress in the kidney is associated with an early systemic γδ T-cell immune cell response.Crit Care. 2018 Jul 4;22(1):168. doi: 10.1186/s13054-018-2094-x. Crit Care. 2018. PMID: 29973233 Free PMC article.
-
Immunomodulatory Effects of Mesenchymal Stem Cells on Drug-Induced Acute Kidney Injury.Front Immunol. 2021 Jun 4;12:683003. doi: 10.3389/fimmu.2021.683003. eCollection 2021. Front Immunol. 2021. PMID: 34149721 Free PMC article. Review.
-
Toll-like Receptor 4 in Acute Kidney Injury.Int J Mol Sci. 2023 Jan 11;24(2):1415. doi: 10.3390/ijms24021415. Int J Mol Sci. 2023. PMID: 36674930 Free PMC article. Review.
References
-
- Wallach D, Kang TB, Kovalenko A: Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat Rev Immunol 14: 51–59, 2014 - PubMed
-
- Barry M, Bleackley RC: Cytotoxic T lymphocytes: All roads lead to death. Nat Rev Immunol 2: 401–409, 2002 - PubMed
-
- Zipfel PF, Skerka C: Complement regulators and inhibitory proteins. Nat Rev Immunol 9: 729–740, 2009 - PubMed
-
- Allam R, Anders HJ: The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol 20: 538–544, 2008 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
