

Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene
- PMID: 26291278
- PMCID: PMC4548837
- DOI: 10.1016/j.bbagrm.2015.08.004
Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene
Abstract
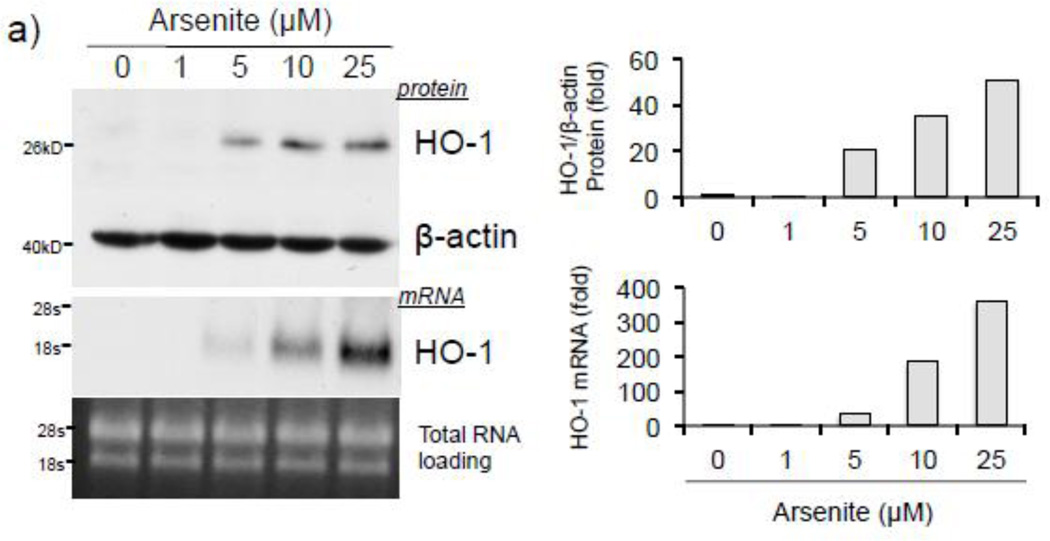
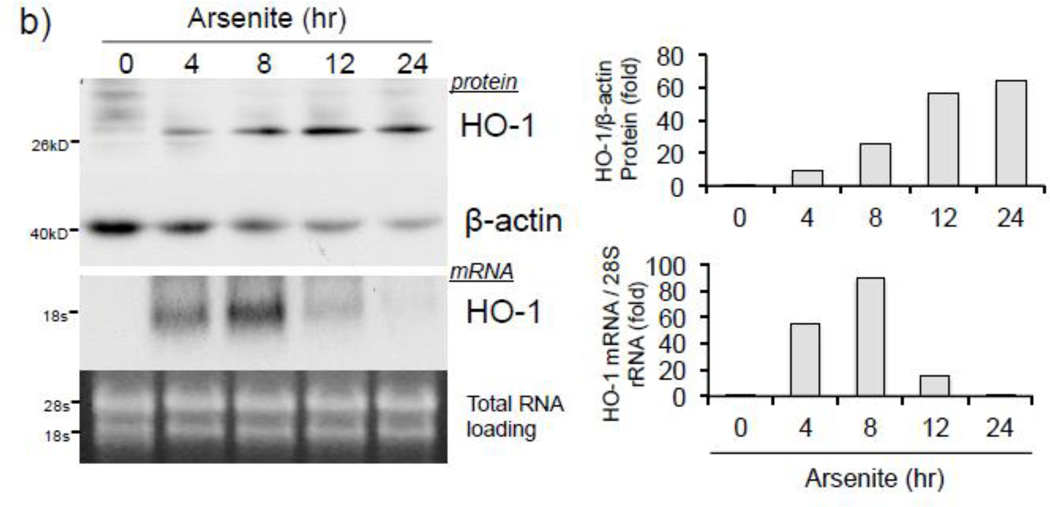
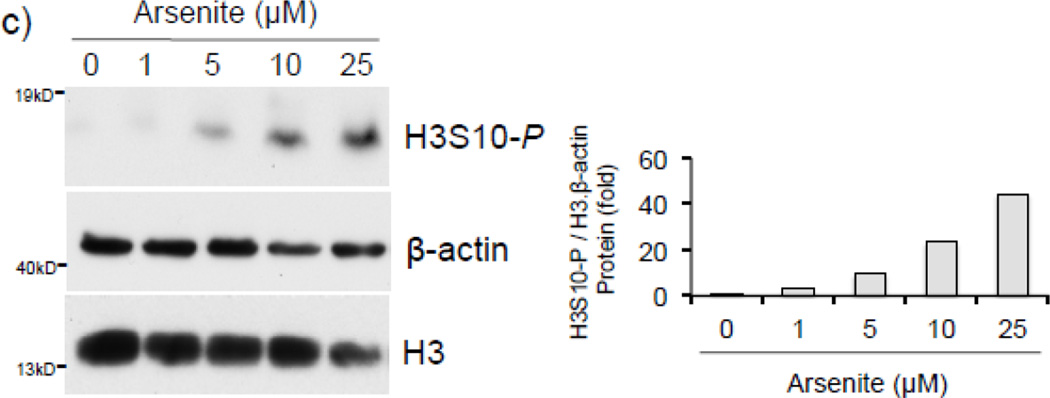
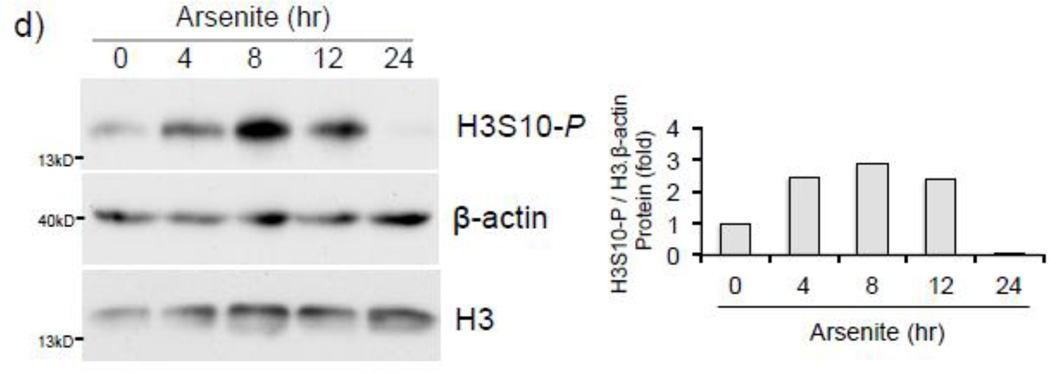
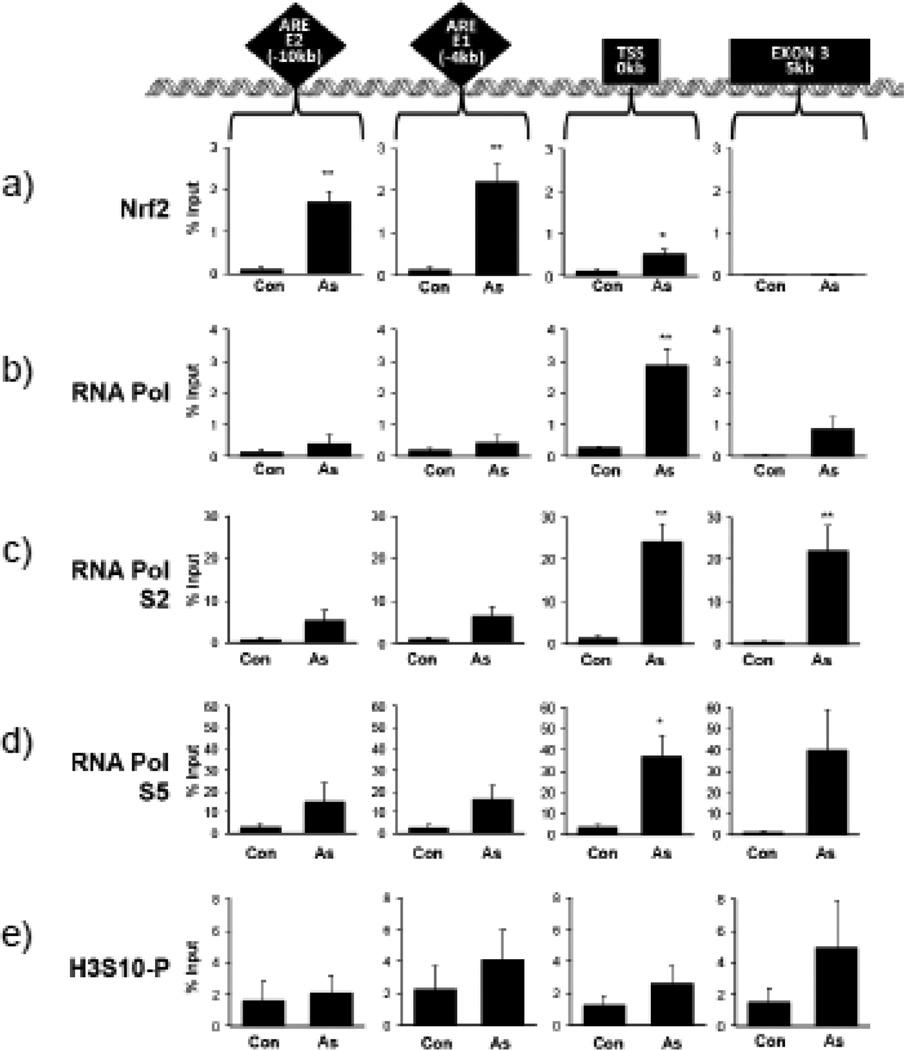
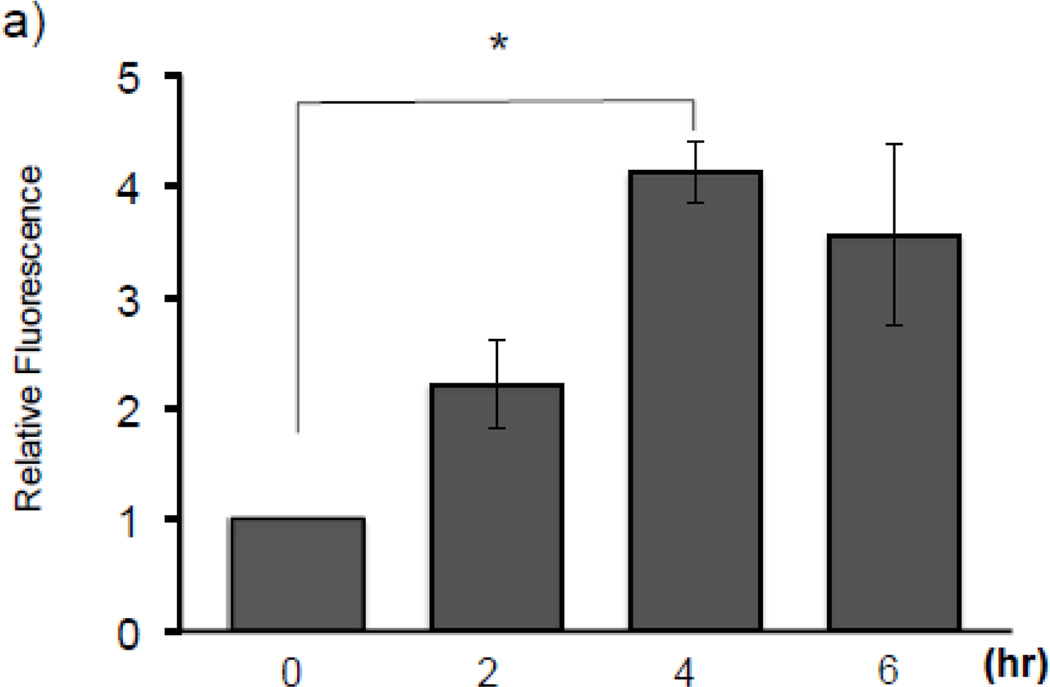
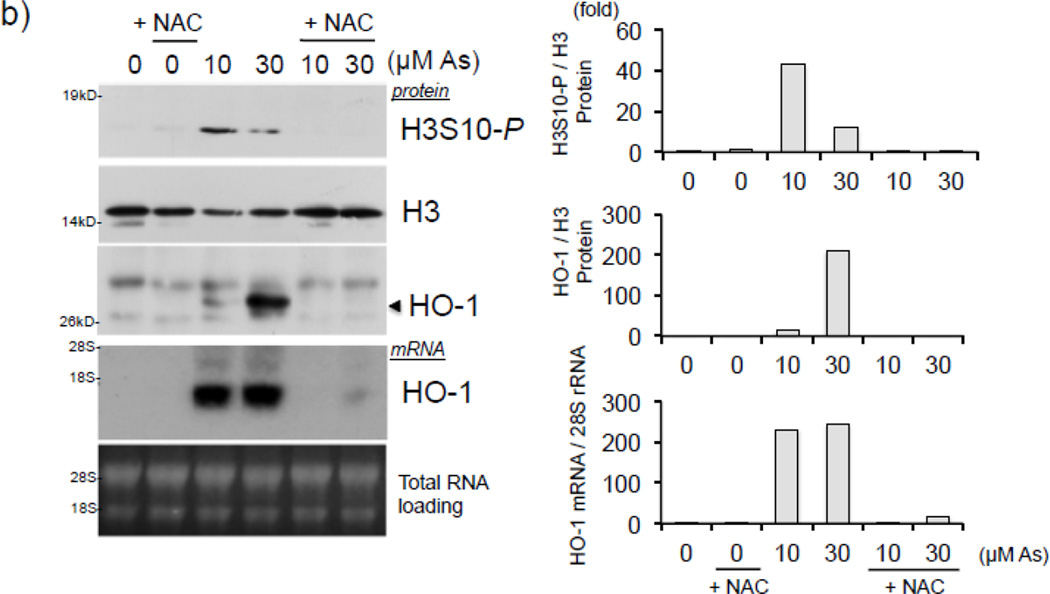
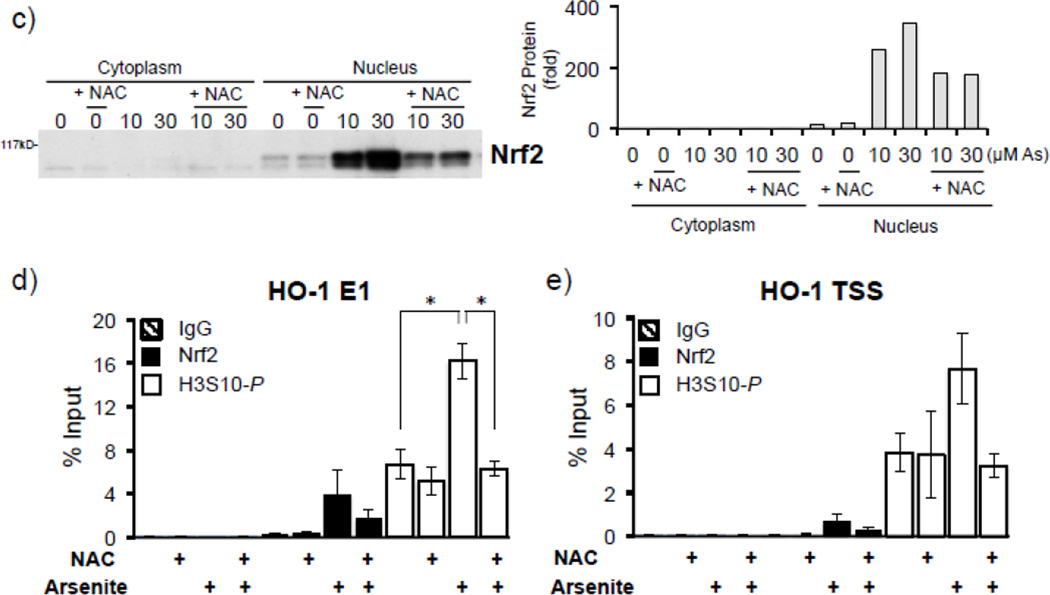
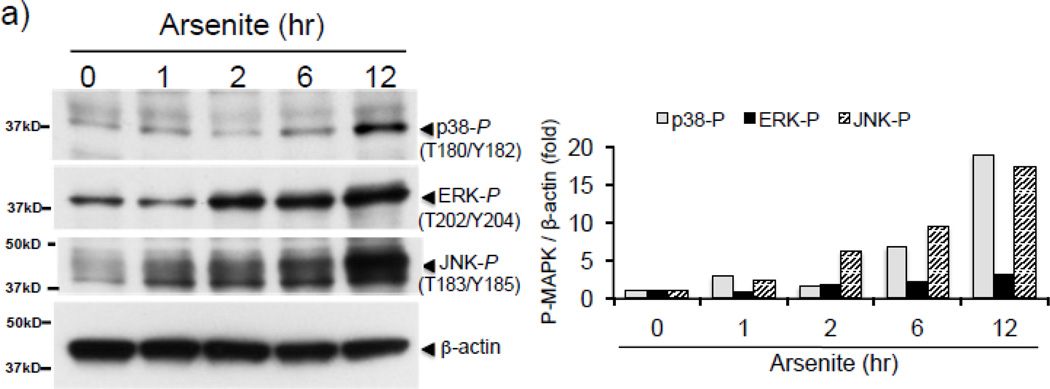
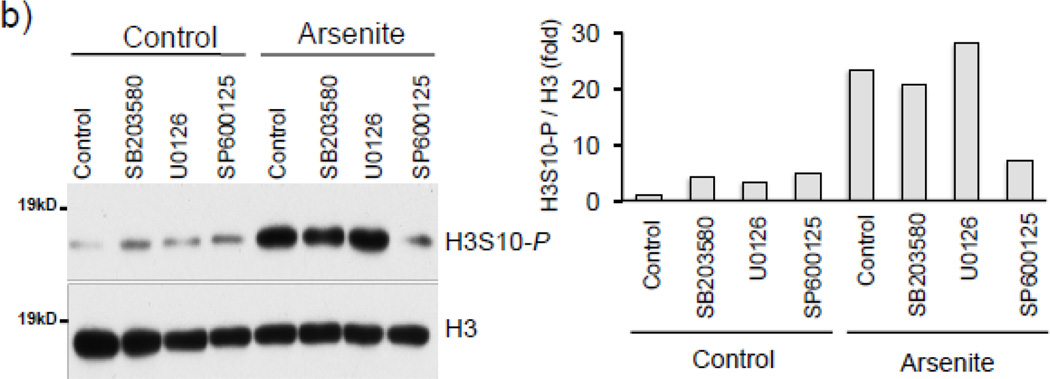
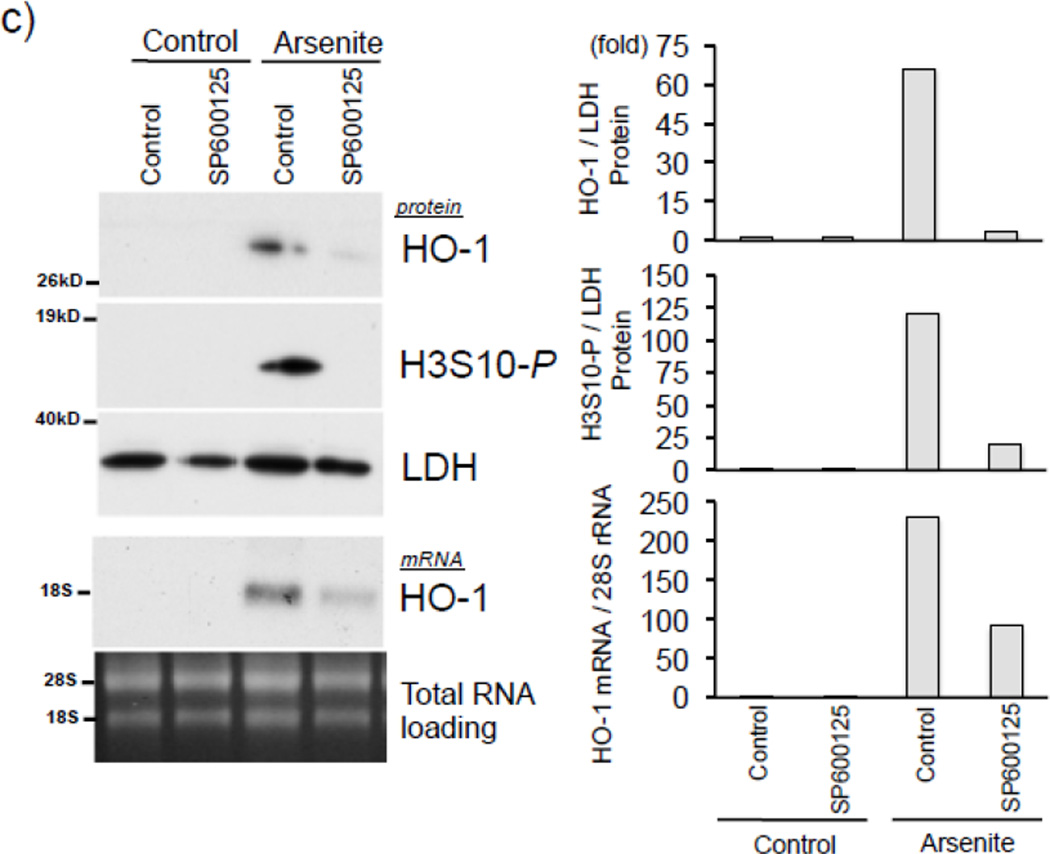
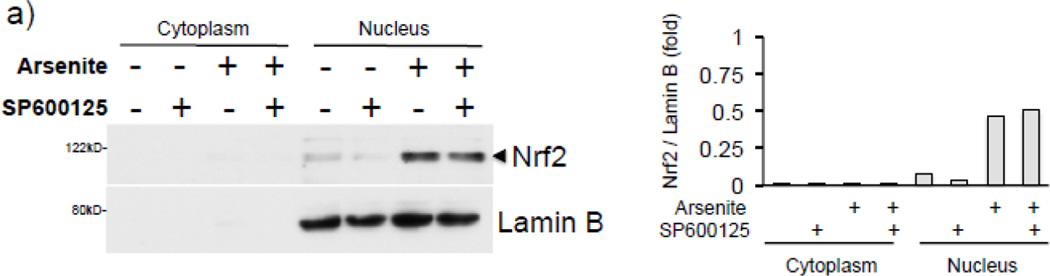
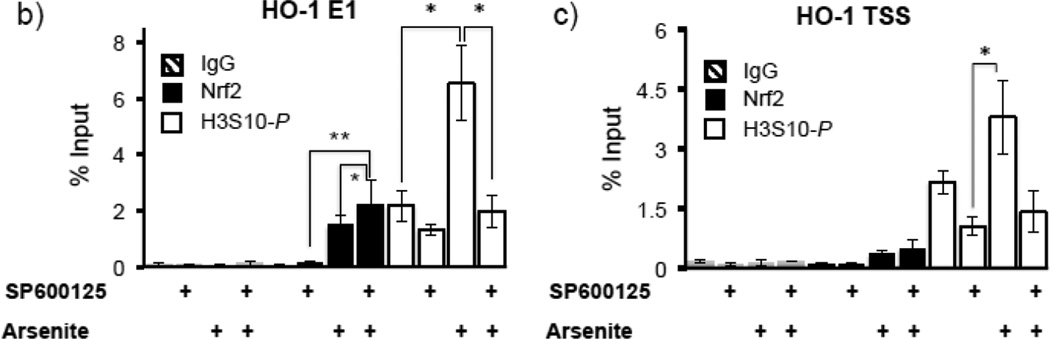
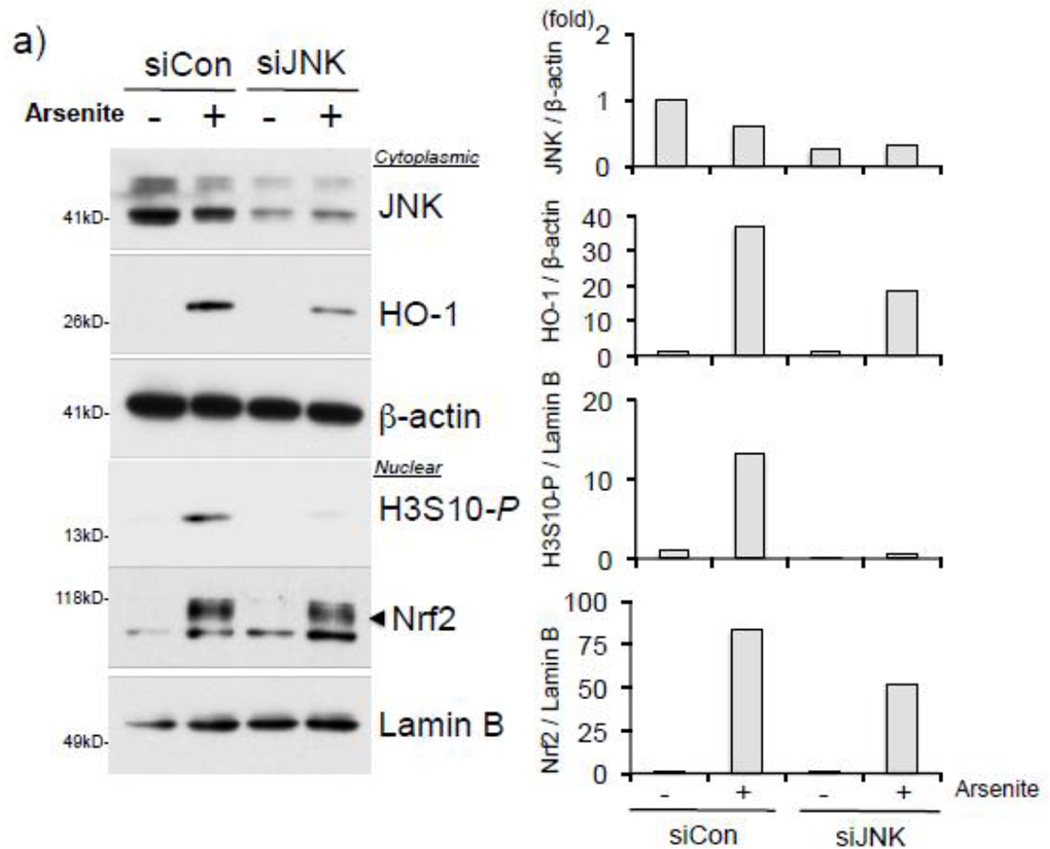
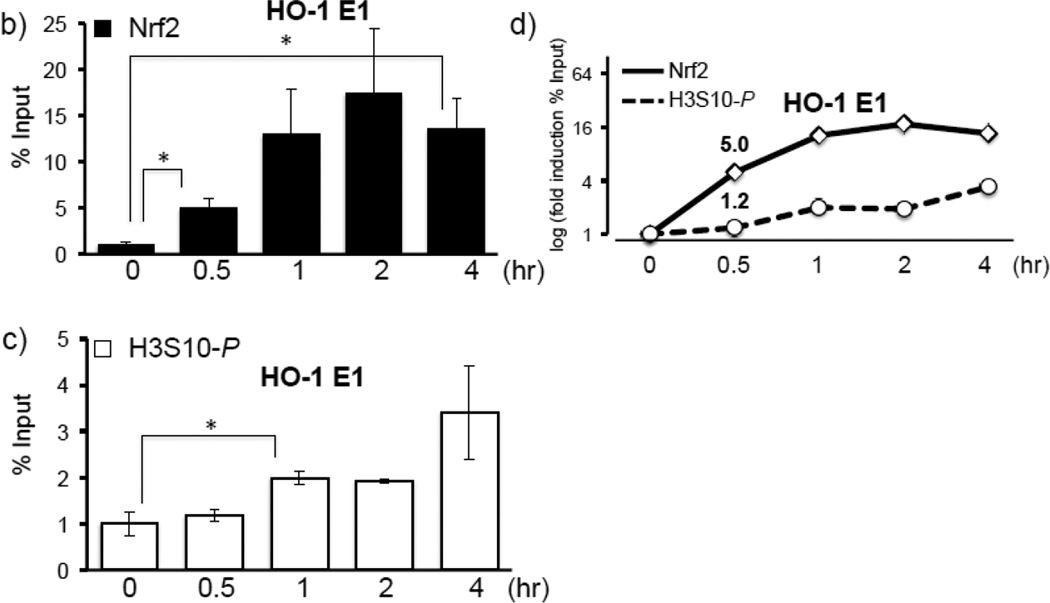
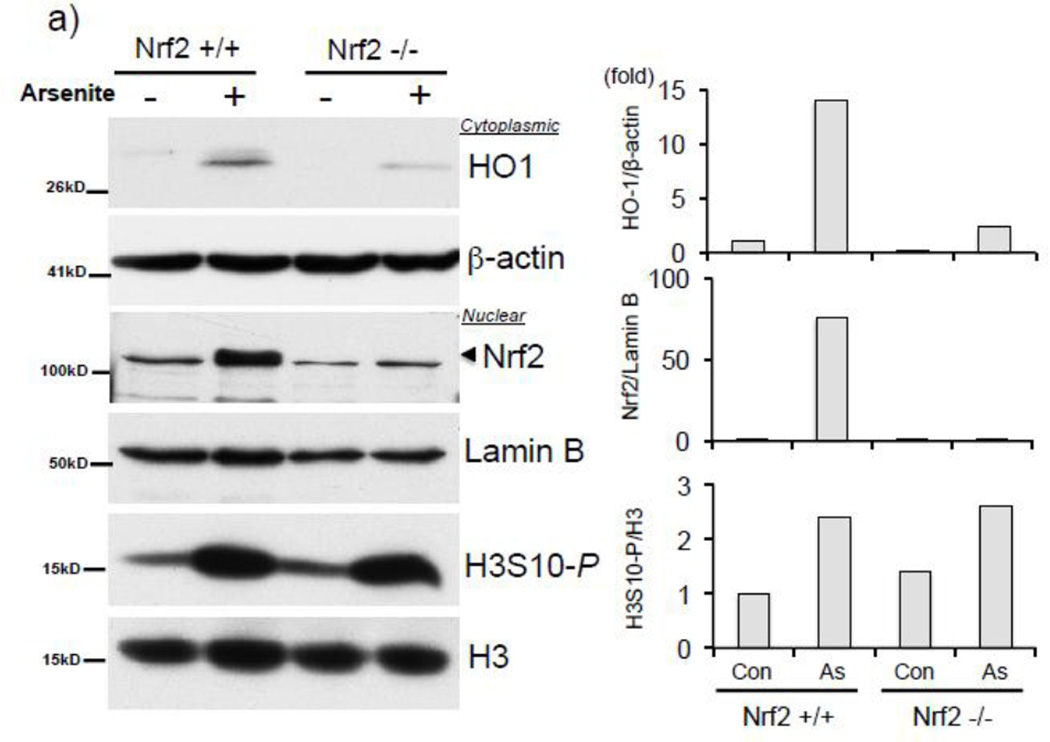
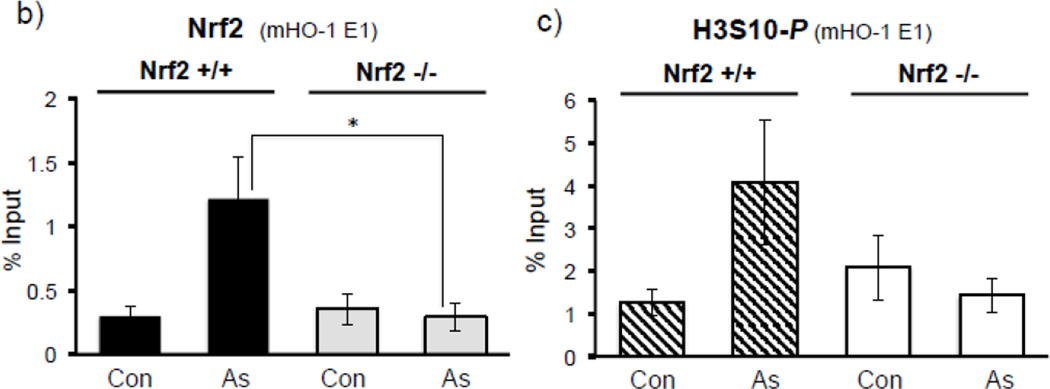
Expression of the antioxidant gene heme oxygenase-1 (HO-1) is primarily induced through NF-E2-related factor 2 (Nrf2)-mediated activation of the antioxidant response element (ARE). Gene transcription is coordinately regulated by transcription factor activity at enhancer elements and epigenetic alterations such as the posttranslational modification of histone proteins. However, the role of histone modifications in the Nrf2-ARE axis remains largely uncharacterized. The environmental contaminant arsenite is a potent inducer of both HO-1 expression and phosphorylation of histone H3 serine 10 (H3S10); therefore, we investigated the relationships between Nrf2 and H3S10 phosphorylation in arsenite-induced, ARE-dependent, transcriptional activation of the human HO-1 gene. Arsenite increased phosphorylation of H3S10 both globally and at the HO-1 promoter concomitantly with HO-1 transcription in human HaCaT keratinocytes. Conversely, arsenite-induced H3S10 phosphorylation and HO-1 expression were blocked by N-acetylcysteine (NAC), the c-Jun N-terminal kinase (JNK) inhibitor SP600125, and JNK knockdown (siJNK). Interestingly, ablation of arsenite-induced H3S10 phosphorylation by SP600125 or siJNK did not inhibit Nrf2 nuclear accumulation nor ARE binding, despite inhibiting HO-1 expression. In response to arsenite, binding of Nrf2 to the HO-1 ARE preceded phosphorylation of H3S10 at the HO-1 ARE. Furthermore, arsenite-mediated occupancy of phosphorylated H3S10 at the HO-1 ARE was decreased in Nrf2-deficient mouse embryonic fibroblasts. These results suggest the involvement of H3S10 phosphorylation in the Nrf2-ARE axis by proposing that Nrf2 may influence H3S10 phosphorylation at the HO-1 ARE and additional promoter regions. Our data highlights the complex interplay between Nrf2 and H3S10 phosphorylation in arsenite-activated HO-1 transcription.
Keywords: Antioxidant response element; Arsenic; Epigenetic; HO-1; Histone; Nrf2.
Copyright © 2015 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Differential roles for Nrf2 and AP-1 in upregulation of HO-1 expression by arsenite in murine embryonic fibroblasts.Free Radic Res. 2008 Apr;42(4):297-304. doi: 10.1080/10715760801975735. Free Radic Res. 2008. PMID: 18404528
-
7,8-Dihydroxyflavone protects human keratinocytes against oxidative stress-induced cell damage via the ERK and PI3K/Akt-mediated Nrf2/HO-1 signaling pathways.Int J Mol Med. 2014 Apr;33(4):964-70. doi: 10.3892/ijmm.2014.1643. Epub 2014 Feb 4. Int J Mol Med. 2014. PMID: 24503931
-
Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway.Cell Signal. 2014 Mar;26(3):512-20. doi: 10.1016/j.cellsig.2013.11.029. Epub 2013 Dec 2. Cell Signal. 2014. PMID: 24308969
-
The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance.Oxid Med Cell Longev. 2016;2016:1958174. doi: 10.1155/2016/1958174. Epub 2015 Nov 30. Oxid Med Cell Longev. 2016. PMID: 26697129 Free PMC article. Review.
-
The Nrf2-HO-1 system and inflammaging.Front Immunol. 2024 Sep 24;15:1457010. doi: 10.3389/fimmu.2024.1457010. eCollection 2024. Front Immunol. 2024. PMID: 39380993 Free PMC article. Review.
Cited by
-
JNK1 ablation in mice confers long-term metabolic protection from diet-induced obesity at the cost of moderate skin oxidative damage.FASEB J. 2016 Sep;30(9):3124-32. doi: 10.1096/fj.201600393R. Epub 2016 May 26. FASEB J. 2016. PMID: 27230858 Free PMC article.
-
Influence of Arsenic on Global Levels of Histone Posttranslational Modifications: a Review of the Literature and Challenges in the Field.Curr Environ Health Rep. 2016 Sep;3(3):225-37. doi: 10.1007/s40572-016-0104-1. Curr Environ Health Rep. 2016. PMID: 27352015 Free PMC article. Review.
-
Quantitative Mass Spectrometry Reveals Changes in Histone H2B Variants as Cells Undergo Inorganic Arsenic-Mediated Cellular Transformation.Mol Cell Proteomics. 2016 Jul;15(7):2411-22. doi: 10.1074/mcp.M116.058412. Epub 2016 May 11. Mol Cell Proteomics. 2016. PMID: 27169413 Free PMC article.
-
Nrf2 Signaling Pathway: Focus on Oxidative Stress in Spinal Cord Injury.Mol Neurobiol. 2025 Feb;62(2):2230-2249. doi: 10.1007/s12035-024-04394-z. Epub 2024 Aug 2. Mol Neurobiol. 2025. PMID: 39093381 Review.
-
Protection of Nrf2 against arsenite-induced oxidative damage is regulated by the cyclic guanosine monophosphate-protein kinase G signaling pathway.Environ Toxicol. 2017 Aug;32(8):2004-2020. doi: 10.1002/tox.22374. Epub 2016 Oct 24. Environ Toxicol. 2017. PMID: 27774770 Free PMC article.
References
-
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M. Arsenic: toxicity, oxidative stress and human disease. Journal of applied toxicology : J. Appl. Tox. 2011;31:95–107. - PubMed
-
- Huang C, Ke Q, Costa M, Shi X. Molecular mechanisms of arsenic carcinogenesis. Mol Cell Biochem. 2004;255:57–66. - PubMed
-
- Shi H, Hudson LG, Ding W, Wang S, Cooper KL, Liu S, Chen Y, Shi X, Liu KJ. Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chem Res Toxicol. 2004;17:871–878. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
