

Global diversity, population stratification, and selection of human copy-number variation
- PMID: 26249230
- PMCID: PMC4568308
- DOI: 10.1126/science.aab3761
Global diversity, population stratification, and selection of human copy-number variation
Abstract
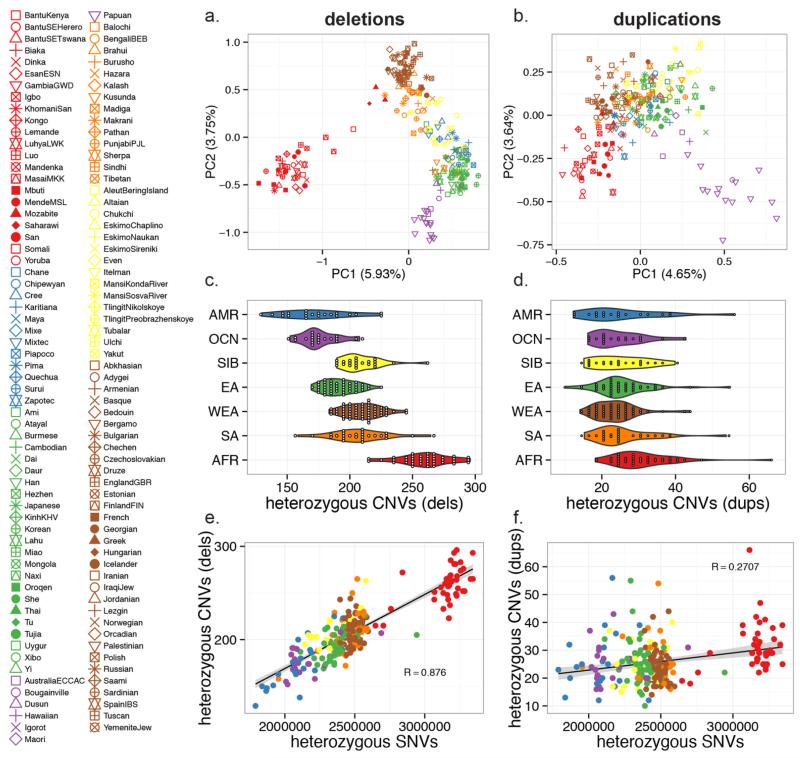
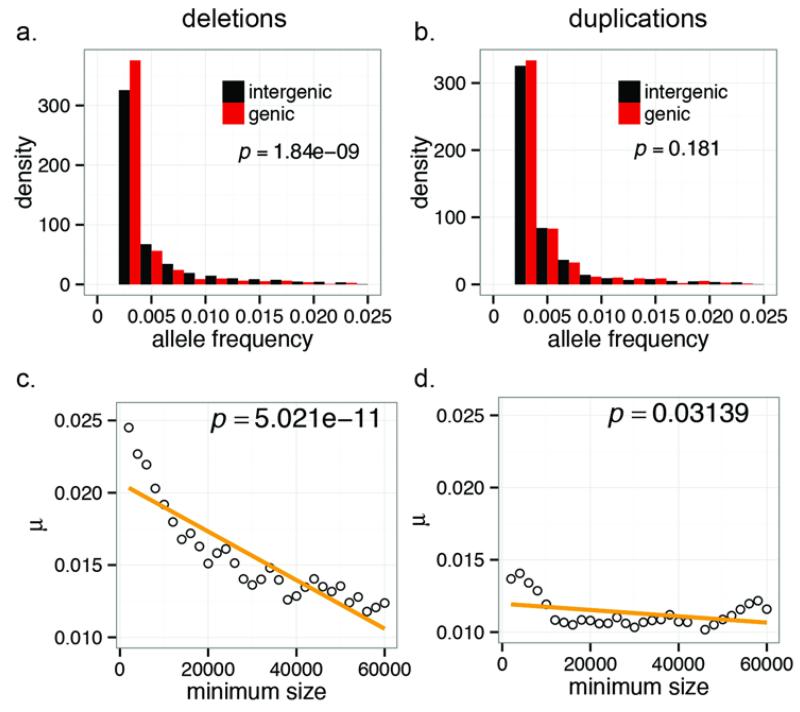
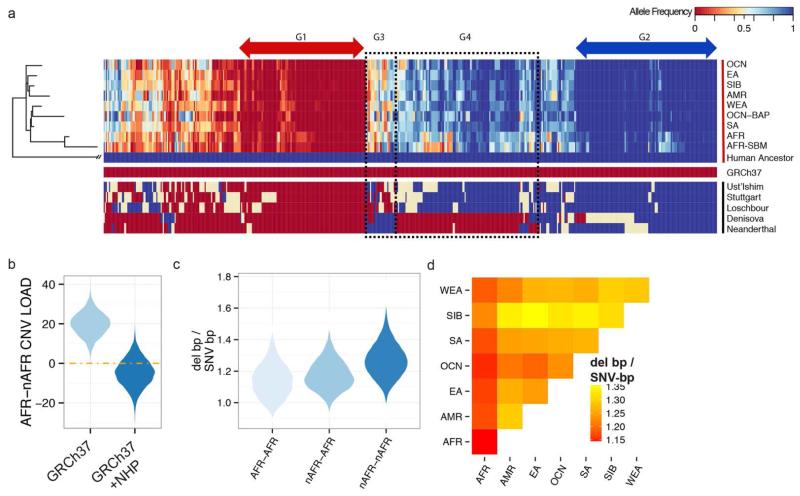
In order to explore the diversity and selective signatures of duplication and deletion human copy-number variants (CNVs), we sequenced 236 individuals from 125 distinct human populations. We observed that duplications exhibit fundamentally different population genetic and selective signatures than deletions and are more likely to be stratified between human populations. Through reconstruction of the ancestral human genome, we identify megabases of DNA lost in different human lineages and pinpoint large duplications that introgressed from the extinct Denisova lineage now found at high frequency exclusively in Oceanic populations. We find that the proportion of CNV base pairs to single-nucleotide-variant base pairs is greater among non-Africans than it is among African populations, but we conclude that this difference is likely due to unique aspects of non-African population history as opposed to differences in CNV load.
Copyright © 2015, American Association for the Advancement of Science.
Figures

Similar articles
-
Evolution and diversity of copy number variation in the great ape lineage.Genome Res. 2013 Sep;23(9):1373-82. doi: 10.1101/gr.158543.113. Epub 2013 Jul 3. Genome Res. 2013. PMID: 23825009 Free PMC article.
-
Copy number variation in human genomes from three major ethno-linguistic groups in Africa.BMC Genomics. 2020 Apr 10;21(1):289. doi: 10.1186/s12864-020-6669-y. BMC Genomics. 2020. PMID: 32272904 Free PMC article.
-
Detecting archaic introgression using an unadmixed outgroup.PLoS Genet. 2018 Sep 18;14(9):e1007641. doi: 10.1371/journal.pgen.1007641. eCollection 2018 Sep. PLoS Genet. 2018. PMID: 30226838 Free PMC article.
-
A copy number variation map of the human genome.Nat Rev Genet. 2015 Mar;16(3):172-83. doi: 10.1038/nrg3871. Epub 2015 Feb 3. Nat Rev Genet. 2015. PMID: 25645873 Review.
-
[Structural variation in the human genome contributes to variation of traits].Tidsskr Nor Laegeforen. 2008 Sep 11;128(17):1951-5. Tidsskr Nor Laegeforen. 2008. PMID: 18787571 Review. Norwegian.
Cited by
-
Copy Number Variation in TAS2R Bitter Taste Receptor Genes: Structure, Origin, and Population Genetics.Chem Senses. 2016 Oct;41(8):649-59. doi: 10.1093/chemse/bjw067. Epub 2016 Jun 23. Chem Senses. 2016. PMID: 27340135 Free PMC article.
-
The p53-inducible CLDN7 regulates colorectal tumorigenesis and has prognostic significance.Neoplasia. 2020 Nov;22(11):590-603. doi: 10.1016/j.neo.2020.09.001. Epub 2020 Sep 28. Neoplasia. 2020. PMID: 32992138 Free PMC article.
-
A high-quality human reference panel reveals the complexity and distribution of genomic structural variants.Nat Commun. 2016 Oct 6;7:12989. doi: 10.1038/ncomms12989. Nat Commun. 2016. PMID: 27708267 Free PMC article.
-
Copy Number Variants in Two Northernmost Cattle Breeds Are Related to Their Adaptive Phenotypes.Genes (Basel). 2022 Sep 6;13(9):1595. doi: 10.3390/genes13091595. Genes (Basel). 2022. PMID: 36140763 Free PMC article.
-
Genomic structural variation: A complex but important driver of human evolution.Am J Biol Anthropol. 2023 Aug;181 Suppl 76(Suppl 76):118-144. doi: 10.1002/ajpa.24713. Epub 2023 Feb 16. Am J Biol Anthropol. 2023. PMID: 36794631 Free PMC article. Review.
References
-
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, Alkan C, Kidd JM, Sun Y, Drautz DI, Bouffard P, Muzny DM, Reid JG, Nazareth LV, Wang Q, Burhans R, Riemer C, Wittekindt NE, Moorjani P, Tindall EA, Danko CG, Teo WS, Buboltz AM, Zhang Z, Ma Q, Oosthuysen A, Steenkamp AW, Oostuisen H, Venter P, Gajewski J, Zhang Y, Pugh BF, Makova KD, Nekrutenko A, Mardis ER, Patterson N, Pringle TH, Chiaromonte F, Mullikin JC, Eichler EE, Hardison RC, Gibbs RA, Harkins TT, Hayes VM. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. Medline doi:10.1038/nature08795. - PMC - PubMed
-
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, Lema G, Nyambo TB, Omar SA, Bodo JM, Froment A, Donnelly MP, Kidd KK, Tishkoff SA, Eichler EE. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet. 2012;44:872–880. Medline doi:10.1038/ng.2335. - PMC - PubMed
-
- Gravel S, Zakharia F, Moreno-Estrada A, Byrnes JK, Muzzio M, Rodriguez-Flores JL, Kenny EE, Gignoux CR, Maples BK, Guiblet W, Dutil J, Via M, Sandoval K, Bedoya G, Oleksyk TK, Ruiz-Linares A, Burchard EG, Martinez-Cruzado JC, Bustamante CD. 1000 Genomes Project, Reconstructing Native American migrations from whole-genome and whole-exome data. PLOS Genet. 2013;9:e1004023. Medline. - PMC - PubMed
-
- Raghavan M, DeGiorgio M, Albrechtsen A, Moltke I, Skoglund P, Korneliussen TS, Grønnow B, Appelt M, Gulløv HC, Friesen TM, Fitzhugh W, Malmström H, Rasmussen S, Olsen J, Melchior L, Fuller BT, Fahrni SM, Stafford T, Jr., Grimes V, Renouf MA, Cybulski J, Lynnerup N, Lahr MM, Britton K, Knecht R, Arneborg J, Metspalu M, Cornejo OE, Malaspinas AS, Wang Y, Rasmussen M, Raghavan V, Hansen TV, Khusnutdinova E, Pierre T, Dneprovsky K, Andreasen C, Lange H, Hayes MG, Coltrain J, Spitsyn VA, Götherström A, Orlando L, Kivisild T, Villems R, Crawford MH, Nielsen FC, Dissing J, Heinemeier J, Meldgaard M, Bustamante C, O’Rourke DH, Jakobsson M, Gilbert MT, Nielsen R, Willerslev E. The genetic prehistory of the New World Arctic. Science. 2014;345:1255832. Medline doi:10.1126/science.1255832. - PubMed
-
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Z, Gusic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PL, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Pääbo S. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. Medline doi:10.1126/science.1188021. - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
- 5DP1ES022577 05/DP/NCCDPHP CDC HHS/United States
- 261213/ERC_/European Research Council/International
- 1R01DK104339-01/DK/NIDDK NIH HHS/United States
- R01 DK104339/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 HL119577/HL/NHLBI NIH HHS/United States
- 1R01GM113657-01/GM/NIGMS NIH HHS/United States
- P30 ES013508/ES/NIEHS NIH HHS/United States
- 098051/WT_/Wellcome Trust/United Kingdom
- ImNIH/Intramural NIH HHS/United States
- R01 GM113657/GM/NIGMS NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- DP1 ES022577/ES/NIEHS NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- T32 GM007266/GM/NIGMS NIH HHS/United States
- HHSN26120080001E/PHS HHS/International
- 2R01HG002385/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
