

c-Myc and AMPK Control Cellular Energy Levels by Cooperatively Regulating Mitochondrial Structure and Function
- PMID: 26230505
- PMCID: PMC4521957
- DOI: 10.1371/journal.pone.0134049
c-Myc and AMPK Control Cellular Energy Levels by Cooperatively Regulating Mitochondrial Structure and Function
Abstract
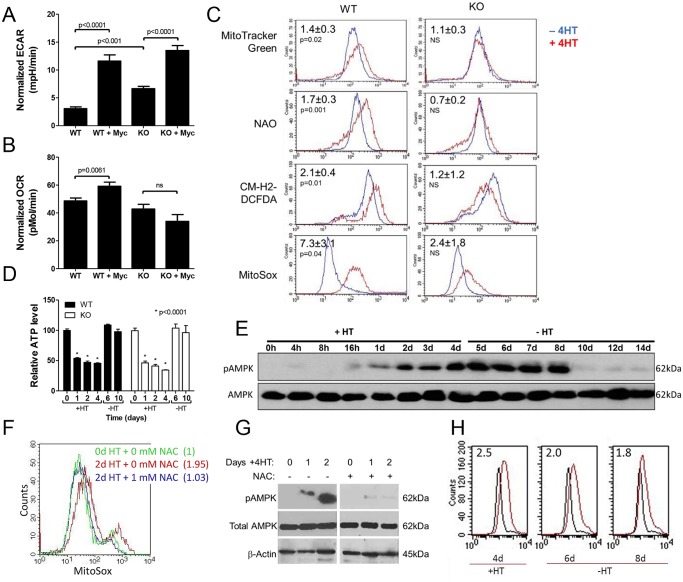
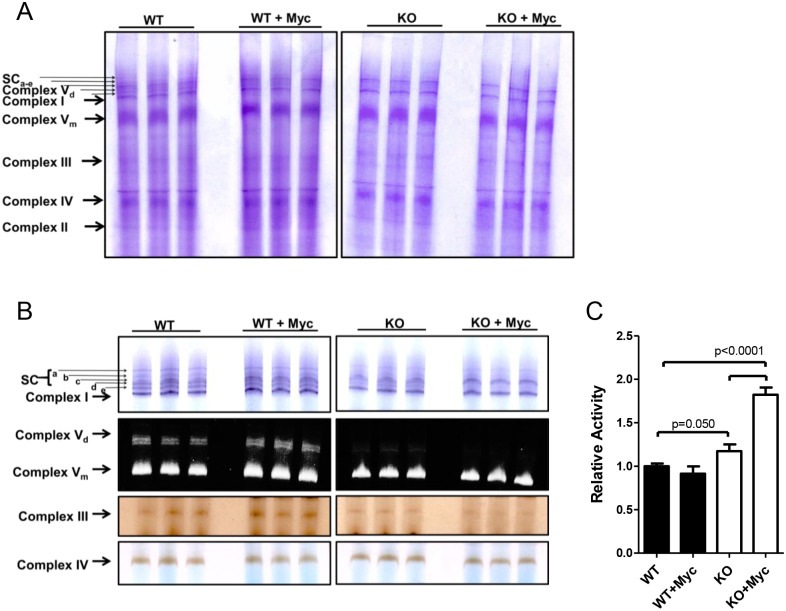
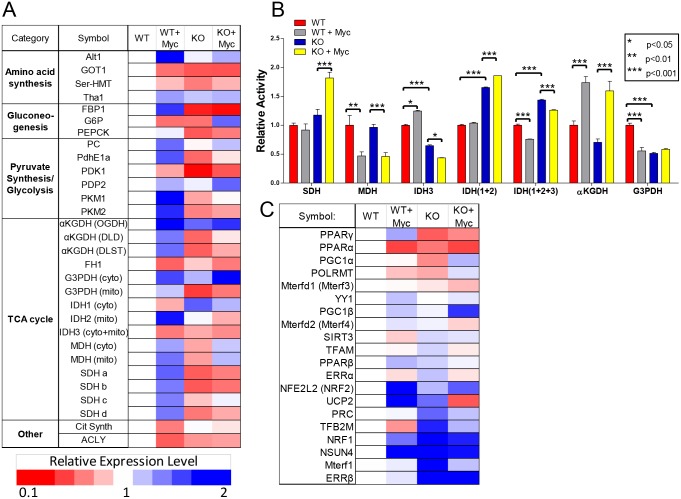
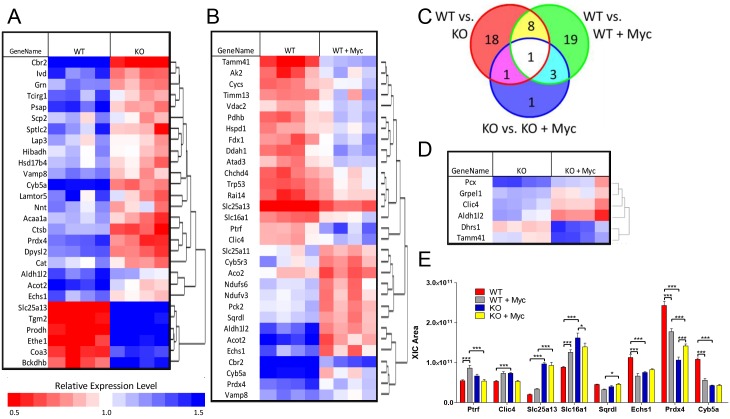
The c-Myc (Myc) oncoprotein and AMP-activated protein kinase (AMPK) regulate glycolysis and oxidative phosphorylation (Oxphos) although often for different purposes. Because Myc over-expression depletes ATP with the resultant activation of AMPK, we explored the potential co-dependency of and cross-talk between these proteins by comparing the consequences of acute Myc induction in ampk+/+ (WT) and ampk-/- (KO) murine embryo fibroblasts (MEFs). KO MEFs showed a higher basal rate of glycolysis than WT MEFs and an appropriate increase in response to activation of a Myc-estrogen receptor (MycER) fusion protein. However, KO MEFs had a diminished ability to increase Oxphos, mitochondrial mass and reactive oxygen species in response to MycER activation. Other differences between WT and KO MEFs, either in the basal state or following MycER induction, included abnormalities in electron transport chain function, levels of TCA cycle-related oxidoreductases and cytoplasmic and mitochondrial redox states. Transcriptional profiling of pathways pertinent to glycolysis, Oxphos and mitochondrial structure and function also uncovered significant differences between WT and KO MEFs and their response to MycER activation. Finally, an unbiased mass-spectrometry (MS)-based survey capable of quantifying ~40% of all mitochondrial proteins, showed about 15% of them to be AMPK- and/or Myc-dependent in their steady state. Significant differences in the activities of the rate-limiting enzymes pyruvate kinase and pyruvate dehydrogenase, which dictate pyruvate and acetyl coenzyme A abundance, were also differentially responsive to Myc and AMPK and could account for some of the differences in basal metabolite levels that were also detected by MS. Thus, Myc and AMPK are highly co-dependent and appear to engage in significant cross-talk across numerous pathways which support metabolic and ATP-generating functions.
Conflict of interest statement
Figures
Similar articles
-
Metabolic and oncogenic adaptations to pyruvate dehydrogenase inactivation in fibroblasts.J Biol Chem. 2019 Apr 5;294(14):5466-5486. doi: 10.1074/jbc.RA118.005200. Epub 2019 Feb 12. J Biol Chem. 2019. PMID: 30755479 Free PMC article.
-
c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate.J Biol Chem. 2014 Sep 5;289(36):25382-92. doi: 10.1074/jbc.M114.580662. Epub 2014 Jul 22. J Biol Chem. 2014. PMID: 25053415 Free PMC article.
-
Mitochondrial AIF loss causes metabolic reprogramming, caspase-independent cell death blockade, embryonic lethality, and perinatal hydrocephalus.Mol Metab. 2020 Oct;40:101027. doi: 10.1016/j.molmet.2020.101027. Epub 2020 May 30. Mol Metab. 2020. PMID: 32480041 Free PMC article.
-
Could MYC induction of mitochondrial biogenesis be linked to ROS production and genomic instability?Cell Cycle. 2005 Nov;4(11):1465-6. doi: 10.4161/cc.4.11.2121. Epub 2005 Nov 23. Cell Cycle. 2005. PMID: 16205115 Review.
-
MYC and AMPK-Save Energy or Die!Front Cell Dev Biol. 2017 Apr 11;5:38. doi: 10.3389/fcell.2017.00038. eCollection 2017. Front Cell Dev Biol. 2017. PMID: 28443281 Free PMC article. Review.
Cited by
-
Metabolic and oncogenic adaptations to pyruvate dehydrogenase inactivation in fibroblasts.J Biol Chem. 2019 Apr 5;294(14):5466-5486. doi: 10.1074/jbc.RA118.005200. Epub 2019 Feb 12. J Biol Chem. 2019. PMID: 30755479 Free PMC article.
-
Preservation of microvascular barrier function requires CD31 receptor-induced metabolic reprogramming.Nat Commun. 2020 Jul 17;11(1):3595. doi: 10.1038/s41467-020-17329-8. Nat Commun. 2020. PMID: 32681081 Free PMC article.
-
Regulation of metastatic potential by drug repurposing and mitochondrial targeting in colorectal cancer cells.BMC Cancer. 2024 Mar 8;24(1):323. doi: 10.1186/s12885-024-12064-5. BMC Cancer. 2024. PMID: 38459456 Free PMC article.
-
Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma.J Biol Chem. 2017 Jun 16;292(24):10068-10086. doi: 10.1074/jbc.M117.782052. Epub 2017 Apr 21. J Biol Chem. 2017. PMID: 28432125 Free PMC article.
-
Regulation of Normal and Neoplastic Proliferation and Metabolism by the Extended Myc Network.Cells. 2022 Dec 8;11(24):3974. doi: 10.3390/cells11243974. Cells. 2022. PMID: 36552737 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
