

Alström syndrome: current perspectives
- PMID: 26229500
- PMCID: PMC4516341
- DOI: 10.2147/TACG.S56612
Alström syndrome: current perspectives
Abstract
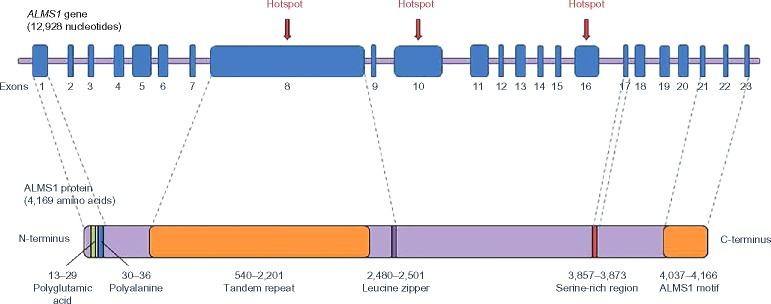
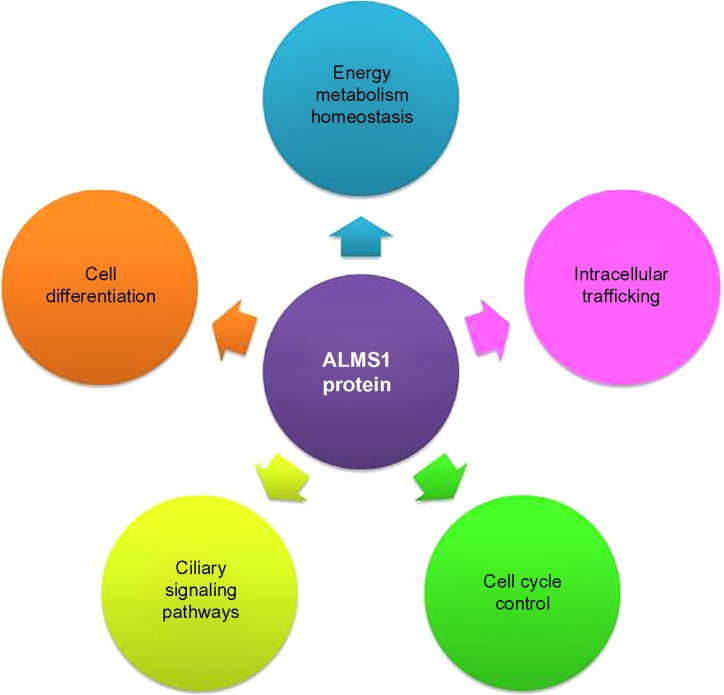
Alström syndrome (ALMS) is a rare genetic disorder that has been included in the ciliopathies group, in the last few years. Ciliopathies are a growing group of diseases associated with defects in ciliary structure and function. The development of more powerful genetic approaches has been replaced the strategies to follow for getting a successful molecular diagnosis for these patients, especially for those without the typical ALMS phenotype. In an effort to deepen the understanding of the pathogenesis of ALMS disease, much work has been done, in order to establish the biological implication of ALMS1 protein, which is still being elucidated. In addition to its role in ciliary function and structure maintenance, this protein has been implicated in intracellular trafficking, regulation of cilia signaling pathways, and cellular differentiation, among others. All these progresses will lead to identifying therapeutic targets, thus opening the way to future personalized therapies for human ciliopathies.
Keywords: ALMS1 gene; ALMS1 protein; Alström syndrome; ciliopathies; molecular diagnosis.
Figures
Similar articles
-
A role for Alström syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence.PLoS Genet. 2007 Jan 5;3(1):e8. doi: 10.1371/journal.pgen.0030008. Epub 2006 Nov 30. PLoS Genet. 2007. PMID: 17206865 Free PMC article.
-
Primary Cilium, An Unsung Hero in Maintaining Functional β-cell Population.Yale J Biol Med. 2019 Sep 20;92(3):471-480. eCollection 2019 Sep. Yale J Biol Med. 2019. PMID: 31543709 Free PMC article. Review.
-
ALMS1 and Alström syndrome: a recessive form of metabolic, neurosensory and cardiac deficits.J Mol Med (Berl). 2019 Jan;97(1):1-17. doi: 10.1007/s00109-018-1714-x. Epub 2018 Nov 12. J Mol Med (Berl). 2019. PMID: 30421101 Free PMC article. Review.
-
The Alström syndrome protein, ALMS1, interacts with α-actinin and components of the endosome recycling pathway.PLoS One. 2012;7(5):e37925. doi: 10.1371/journal.pone.0037925. Epub 2012 May 31. PLoS One. 2012. PMID: 22693585 Free PMC article.
-
Alström syndrome: an ultra-rare monogenic disorder as a model for insulin resistance, type 2 diabetes mellitus and obesity.Endocrine. 2021 Mar;71(3):618-625. doi: 10.1007/s12020-021-02643-y. Epub 2021 Feb 10. Endocrine. 2021. PMID: 33566311 Review.
Cited by
-
Recent advances in understanding liver fibrosis: bridging basic science and individualized treatment concepts.F1000Res. 2018 Jun 27;7:F1000 Faculty Rev-921. doi: 10.12688/f1000research.14841.1. eCollection 2018. F1000Res. 2018. PMID: 30002817 Free PMC article. Review.
-
ALMS1 Regulates TGF-β Signaling and Morphology of Primary Cilia.Front Cell Dev Biol. 2021 Feb 1;9:623829. doi: 10.3389/fcell.2021.623829. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33598462 Free PMC article.
-
Whole-exome sequencing revealed a novel mutation of the ALMS1 gene in a Chinese family with Alström syndrome: a case report.BMC Pediatr. 2024 Aug 2;24(1):494. doi: 10.1186/s12887-024-04949-y. BMC Pediatr. 2024. PMID: 39095761 Free PMC article.
-
Mechanisms of Weight Control by Primary Cilia.Mol Cells. 2022 Apr 30;45(4):169-176. doi: 10.14348/molcells.2022.2046. Mol Cells. 2022. PMID: 35387896 Free PMC article. Review.
-
Treatment with PBI-4050 in patients with Alström syndrome: study protocol for a phase 2, single-Centre, single-arm, open-label trial.BMC Endocr Disord. 2018 Nov 26;18(1):88. doi: 10.1186/s12902-018-0315-6. BMC Endocr Disord. 2018. PMID: 30477455 Free PMC article. Clinical Trial.
References
-
- Alström CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence–Moon–Bardet–Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;(129):1–35. - PubMed
-
- Orphanet: an online rare disease and orphan drug data base [homepage on the Internet] Paris: INSERM; 1997. [Accessed March 13, 2015]. [updated Nov 2012]. Available from: http://www.orpha.net/
-
- Titomanlio L, De Brasi D, Buoninconti A, et al. Alström syndrome: intrafamilial phenotypic variability in sibs with a novel nonsense mutation of the ALMS1 gene. Clin Genet. 2004;65(2):156–157. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
