

Phylodynamic analysis of the canine distemper virus hemagglutinin gene
- PMID: 26205391
- PMCID: PMC4513961
- DOI: 10.1186/s12917-015-0491-9
Phylodynamic analysis of the canine distemper virus hemagglutinin gene
Abstract
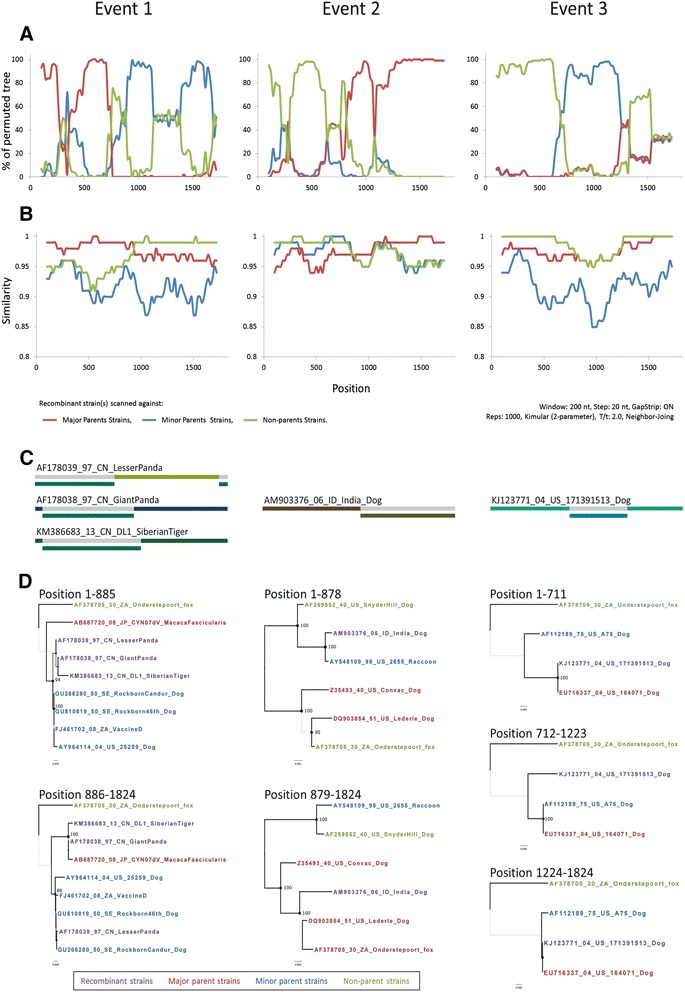
Background: Canine distemper (CD) is one of the most contagious and lethal viral diseases in dogs. Despite the widespread use of vaccines, the prevalence of the CD virus (CDV) has increased at an alarming rate in recent years. In this phylodynamic study, we investigated the spatiotemporal modes of dispersal, viral demographic trends, and effectiveness of vaccines for CDV. A total of 188 full-length CDV hemagglutinin (H) gene sequences dataset were subjected to recombination analysis, including seven from modified live vaccine (MLV) strains and 12 from Taiwan specimens. After excluding the MLV strains and potential recombinant strains, alignments of 176 of 188 previous CDV strains were further used to analyze phylodynamic characteristics, and evidence of selection, and co-evolution.
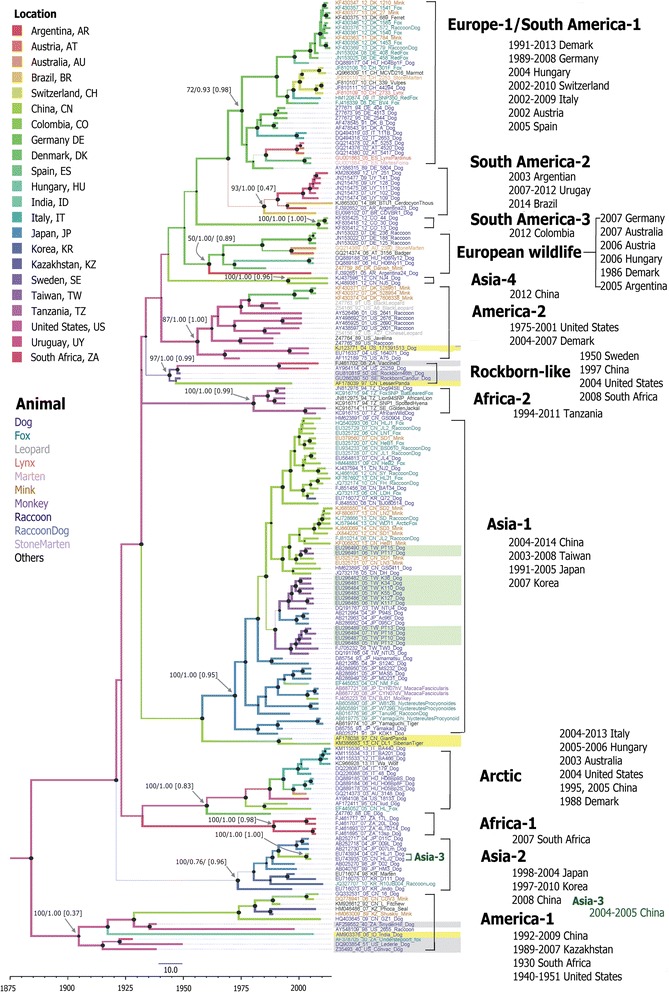
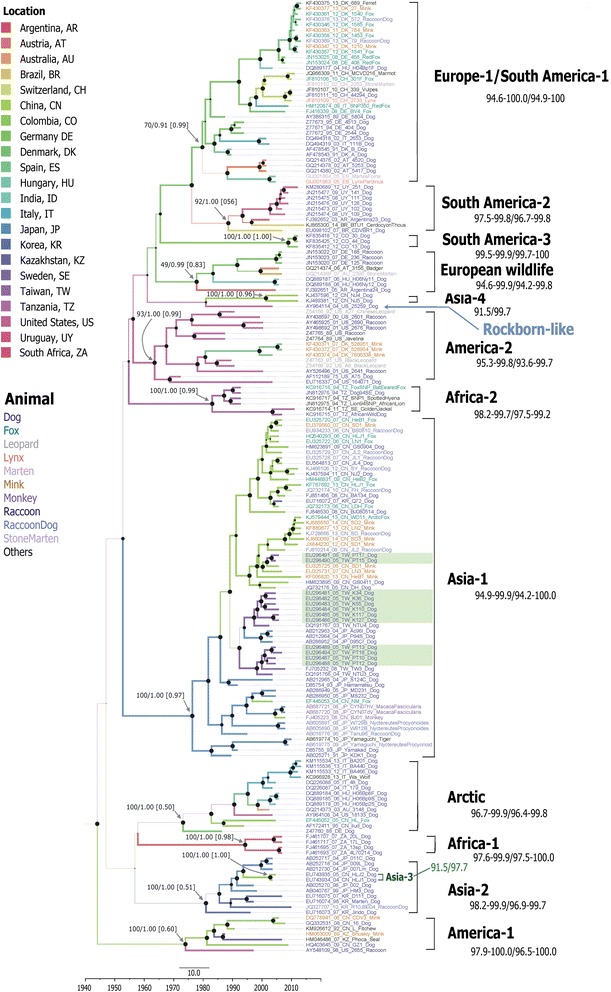
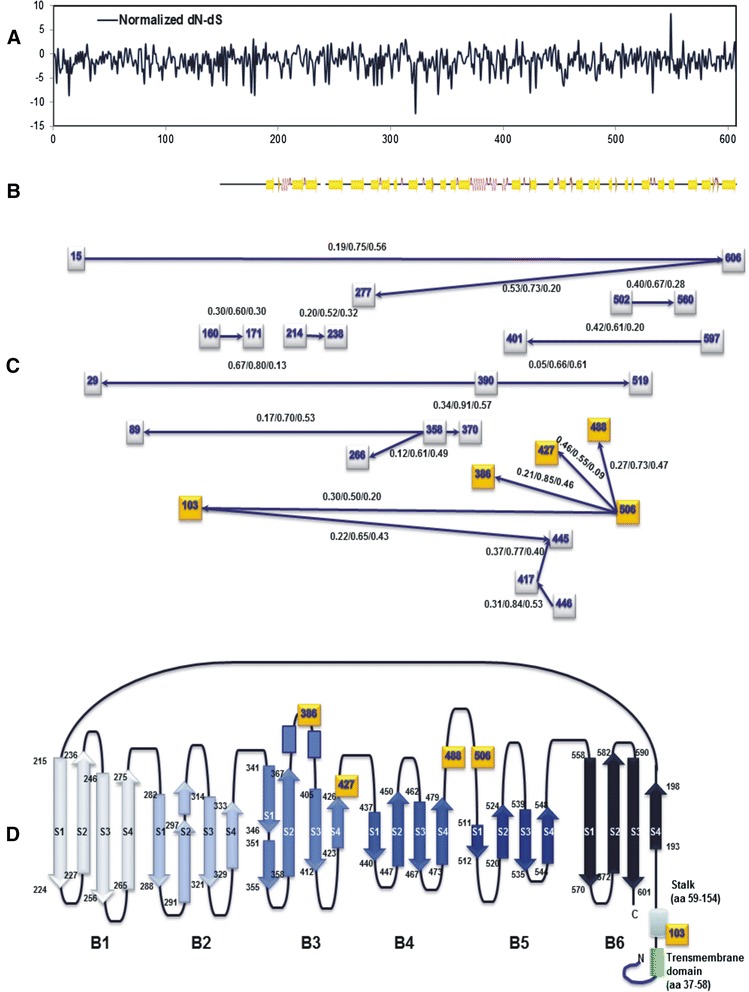
Results: The CDV genotype consisted of MLV-associated genotypes such as America-1 and Rockborn-like strains, which were characterized by long terminal branches and no distinct geographical patterns among lineages. In contrast, wild-type isolates clustered into lineages with a spatiotemporal structure and short terminal branches. Co-circulation and extensive diversification were simultaneously observed. The sequence variation signature was shaped by both geographic diversity and host tropism. Codon 506 was identified as a multi-epistatic interacting in the H protein.
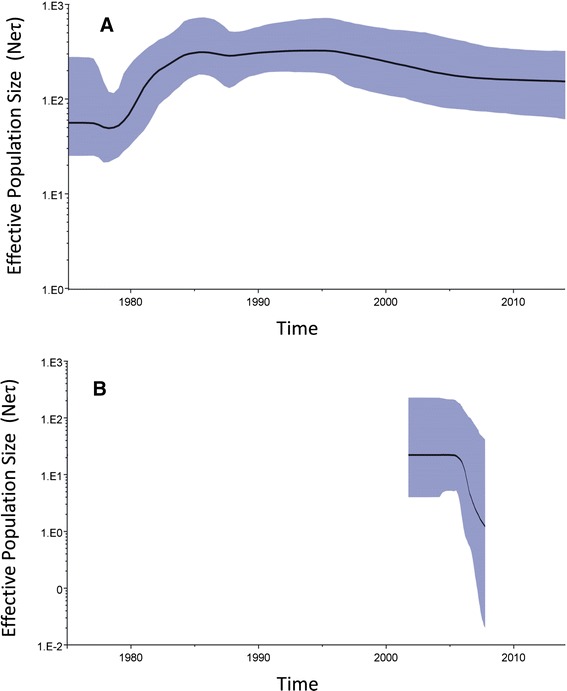
Conclusions: The topological signature revealed in this study suggests different epidemic scenarios. For example, a ladder-like backbone is a hallmark of directional selection, and cladogenesis at long terminal branches indicates the emergence of a surviving lineage. The stable effective viral population of CDV indicate the effectiveness of vaccines currently used to control the virus.
Figures
Similar articles
-
Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy.Vet Microbiol. 2006 Sep 10;116(4):301-9. doi: 10.1016/j.vetmic.2006.04.019. Epub 2006 Apr 27. Vet Microbiol. 2006. PMID: 16730927
-
Phylogenetic analysis of haemagglutinin gene deciphering a new genetically distinct lineage of canine distemper virus circulating among domestic dogs in India.Transbound Emerg Dis. 2019 May;66(3):1252-1267. doi: 10.1111/tbed.13142. Epub 2019 Feb 27. Transbound Emerg Dis. 2019. PMID: 30725534
-
Phylogenetic evidence of a new canine distemper virus lineage among domestic dogs in Colombia, South America.Vet Microbiol. 2014 Aug 6;172(1-2):168-76. doi: 10.1016/j.vetmic.2014.05.019. Epub 2014 May 27. Vet Microbiol. 2014. PMID: 24950886
-
[Genetic variations and cellular receptors of Canine distemper virus--a review].Wei Sheng Wu Xue Bao. 2008 Jul;48(7):986-91. Wei Sheng Wu Xue Bao. 2008. PMID: 18837382 Review. Chinese.
-
Tropism and molecular pathogenesis of canine distemper virus.Virol J. 2019 Mar 7;16(1):30. doi: 10.1186/s12985-019-1136-6. Virol J. 2019. PMID: 30845967 Free PMC article. Review.
Cited by
-
Canine distemper virus N protein induces autophagy to facilitate viral replication.BMC Vet Res. 2023 Mar 15;19(1):60. doi: 10.1186/s12917-023-03575-7. BMC Vet Res. 2023. PMID: 36922800 Free PMC article.
-
Biomolecular Analysis of Canine Distemper Virus Strains in Two Domestic Ferrets (Mustela putorius furo).Vet Sci. 2023 May 26;10(6):375. doi: 10.3390/vetsci10060375. Vet Sci. 2023. PMID: 37368761 Free PMC article.
-
Identification of a new polymorphism on the wild-type canine distemper virus genome: could this contribute to vaccine failures?Braz J Microbiol. 2023 Jun;54(2):665-678. doi: 10.1007/s42770-023-00971-x. Epub 2023 May 4. Braz J Microbiol. 2023. PMID: 37140816 Free PMC article.
-
Phylodynamic analysis of two amino acid substitutions in the hemagglutinin protein of canine distemper virus strains detected in fur-bearing animals in China.Virus Genes. 2020 Feb;56(1):58-66. doi: 10.1007/s11262-019-01720-9. Epub 2019 Dec 4. Virus Genes. 2020. PMID: 31802380
-
Genetic Adaptations, Biases, and Evolutionary Analysis of Canine Distemper Virus Asia-4 Lineage in a Fatal Outbreak of Wild-Caught Civets in Thailand.Viruses. 2020 Mar 26;12(4):361. doi: 10.3390/v12040361. Viruses. 2020. PMID: 32224857 Free PMC article.
References
-
- Murphy FA, Gibbs EPJ, Horzinek MC, Studdert MJ. Veterinary Virology. 3. San Diego, Calif: Academic; 1999.
-
- Blixenkrone-Moller M. Biological properties of phocine distemper virus and canine distemper virus. APMIS Suppl. 1993;36:1–51. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
