

Huntington's disease: Neural dysfunction linked to inositol polyphosphate multikinase
- PMID: 26195796
- PMCID: PMC4534278
- DOI: 10.1073/pnas.1511810112
Huntington's disease: Neural dysfunction linked to inositol polyphosphate multikinase
Abstract
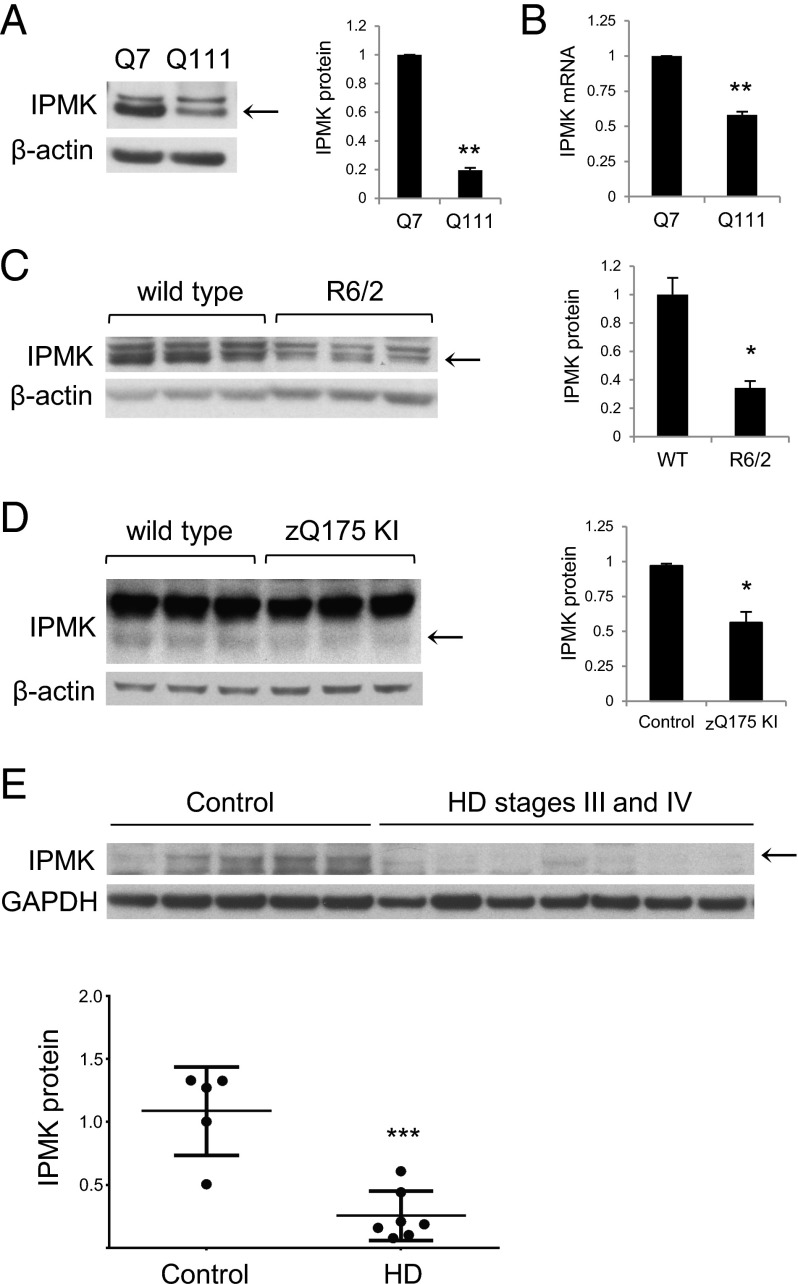
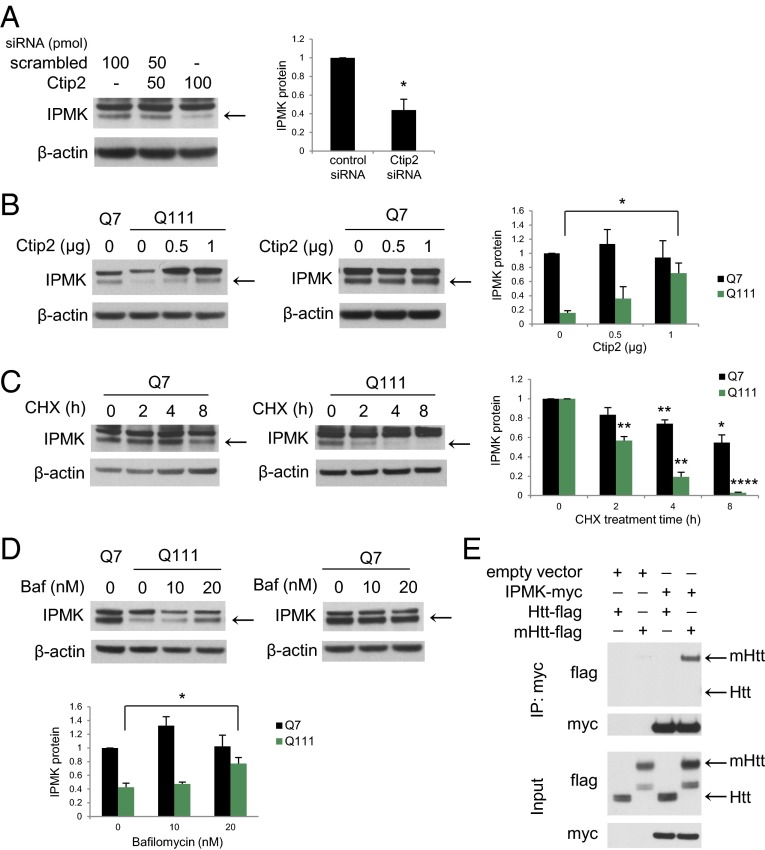
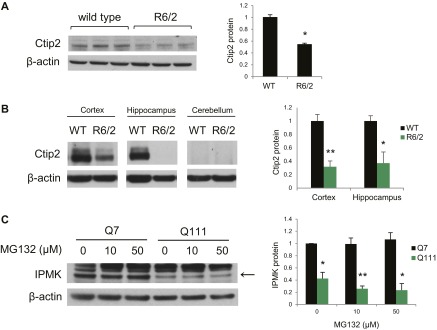
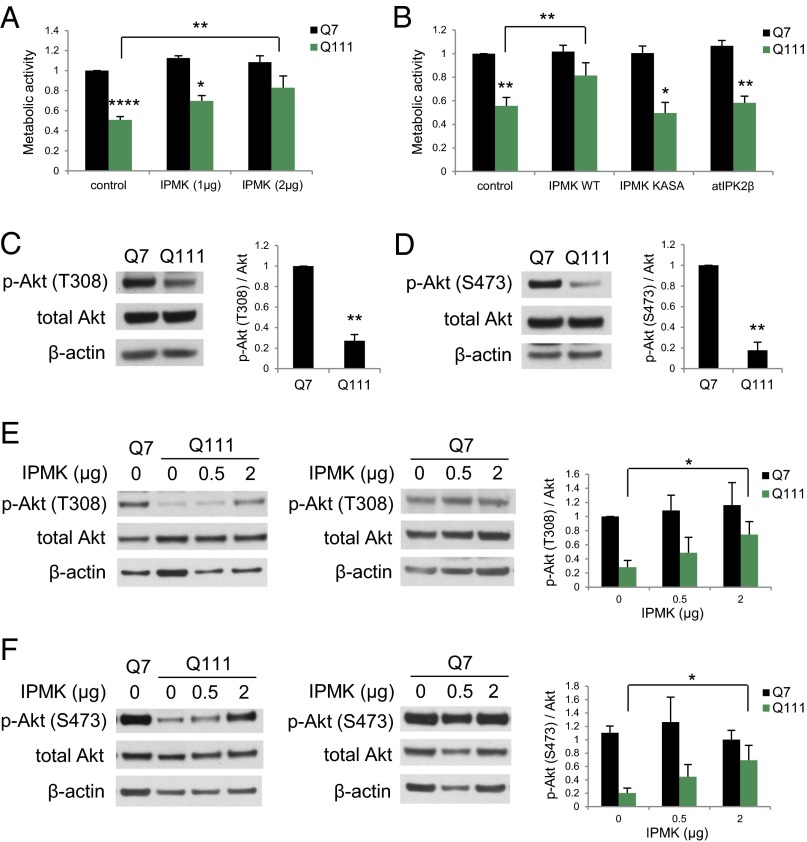
Huntington's disease (HD) is a progressive neurodegenerative disease caused by a glutamine repeat expansion in mutant huntingtin (mHtt). Despite the known genetic cause of HD, the pathophysiology of this disease remains to be elucidated. Inositol polyphosphate multikinase (IPMK) is an enzyme that displays soluble inositol phosphate kinase activity, lipid kinase activity, and various noncatalytic interactions. We report a severe loss of IPMK in the striatum of HD patients and in several cellular and animal models of the disease. This depletion reflects mHtt-induced impairment of COUP-TF-interacting protein 2 (Ctip2), a striatal-enriched transcription factor for IPMK, as well as alterations in IPMK protein stability. IPMK overexpression reverses the metabolic activity deficit in a cell model of HD. IPMK depletion appears to mediate neural dysfunction, because intrastriatal delivery of IPMK abates the progression of motor abnormalities and rescues striatal pathology in transgenic murine models of HD.
Keywords: Akt; Ctip2; Huntington's disease; IPMK; inositol polyphosphate multikinase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

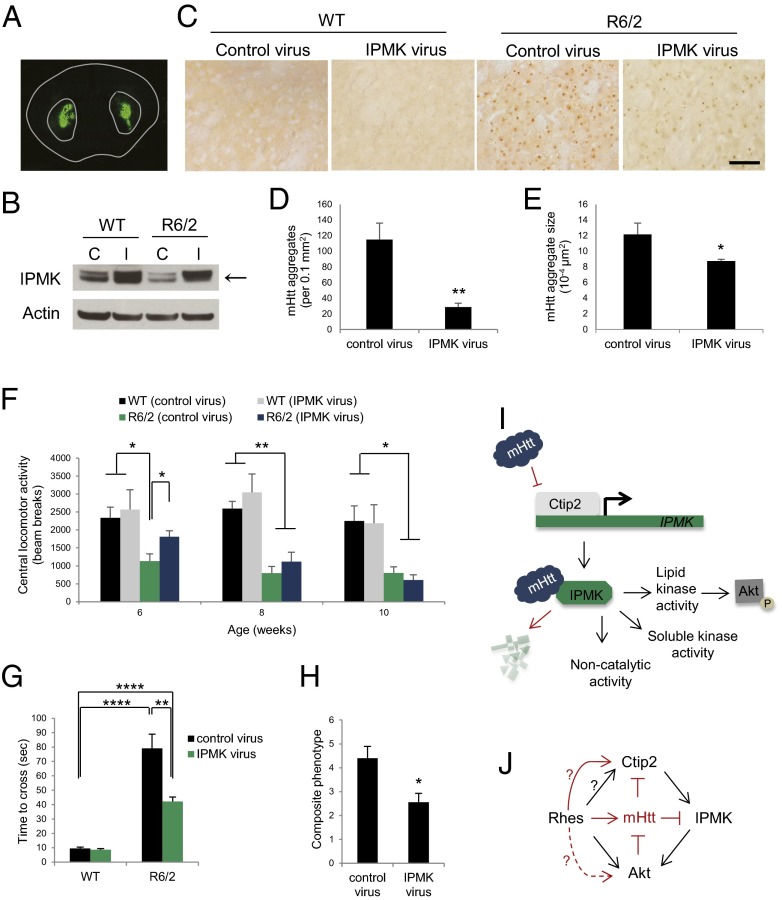
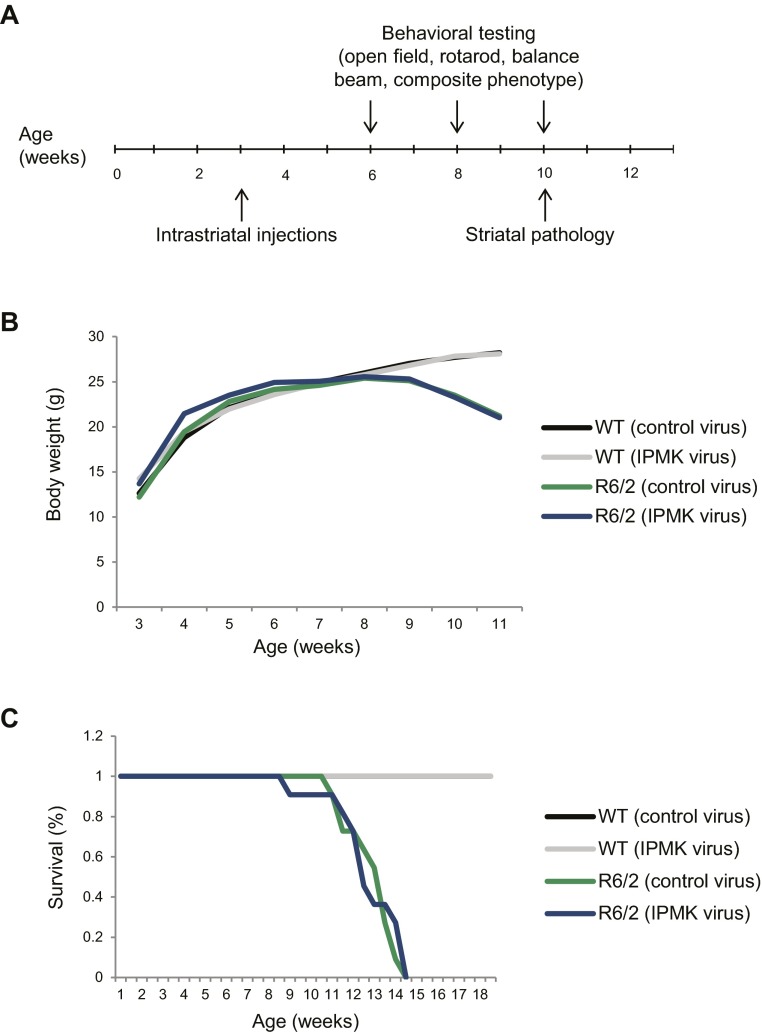
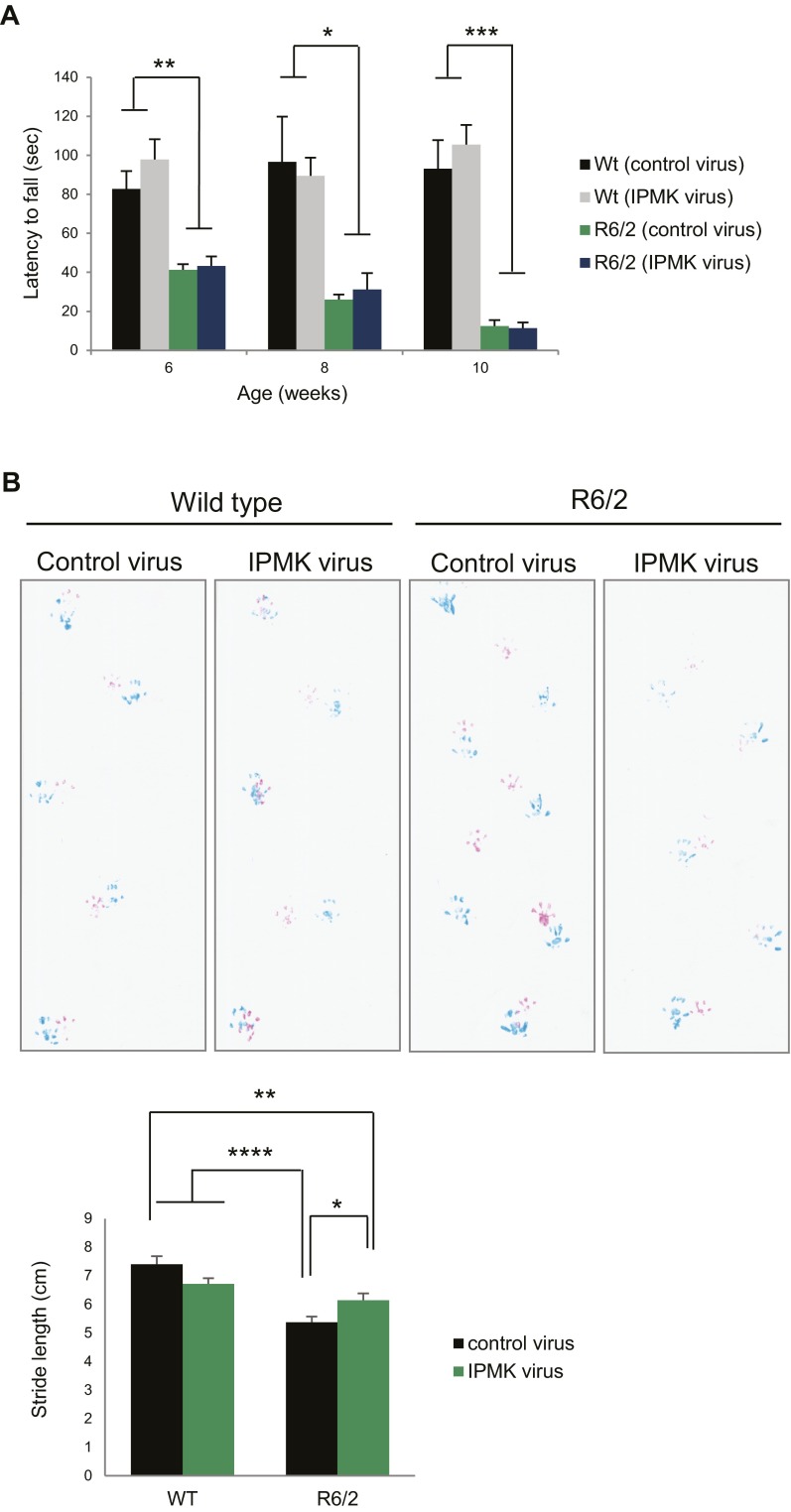
Similar articles
-
The Expanding Significance of Inositol Polyphosphate Multikinase as a Signaling Hub.Mol Cells. 2017 May 31;40(5):315-321. doi: 10.14348/molcells.2017.0066. Epub 2017 May 29. Mol Cells. 2017. PMID: 28554203 Free PMC article. Review.
-
Inositol polyphosphate multikinase (IPMK) in transcriptional regulation and nuclear inositide metabolism.Biochem Soc Trans. 2016 Feb;44(1):279-85. doi: 10.1042/BST20150225. Biochem Soc Trans. 2016. PMID: 26862216 Free PMC article. Review.
-
Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB.Proc Natl Acad Sci U S A. 2011 Jan 25;108(4):1391-6. doi: 10.1073/pnas.1017831108. Epub 2011 Jan 10. Proc Natl Acad Sci U S A. 2011. PMID: 21220345 Free PMC article.
-
Relationship between BDNF expression in major striatal afferents, striatum morphology and motor behavior in the R6/2 mouse model of Huntington's disease.Genes Brain Behav. 2013 Feb;12(1):108-24. doi: 10.1111/j.1601-183X.2012.00858.x. Epub 2012 Nov 21. Genes Brain Behav. 2013. PMID: 23006318
-
AKT-sensitive or insensitive pathways of toxicity in glial cells and neurons in Drosophila models of Huntington's disease.Hum Mol Genet. 2008 Mar 15;17(6):882-94. doi: 10.1093/hmg/ddm360. Epub 2007 Dec 8. Hum Mol Genet. 2008. PMID: 18065778
Cited by
-
The Role of Bcl11 Transcription Factors in Neurodevelopmental Disorders.Biology (Basel). 2024 Feb 17;13(2):126. doi: 10.3390/biology13020126. Biology (Basel). 2024. PMID: 38392344 Free PMC article. Review.
-
Kcs1 and Vip1: The Key Enzymes behind Inositol Pyrophosphate Signaling in Saccharomyces cerevisiae.Biomolecules. 2024 Jan 26;14(2):152. doi: 10.3390/biom14020152. Biomolecules. 2024. PMID: 38397389 Free PMC article. Review.
-
Neuroprotective effect of caffeic acid phenethyl ester in 3-nitropropionic acid-induced striatal neurotoxicity.Korean J Physiol Pharmacol. 2016 May;20(3):279-86. doi: 10.4196/kjpp.2016.20.3.279. Epub 2016 Apr 26. Korean J Physiol Pharmacol. 2016. PMID: 27162482 Free PMC article.
-
Evidence of TAF1 dysfunction in peripheral models of X-linked dystonia-parkinsonism.Cell Mol Life Sci. 2016 Aug;73(16):3205-15. doi: 10.1007/s00018-016-2159-4. Epub 2016 Feb 15. Cell Mol Life Sci. 2016. PMID: 26879577 Free PMC article.
-
Quantification of Huntington's Disease Related Markers in the R6/2 Mouse Model.Front Mol Neurosci. 2021 Jan 11;13:617229. doi: 10.3389/fnmol.2020.617229. eCollection 2020. Front Mol Neurosci. 2021. PMID: 33505246 Free PMC article.
References
-
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. - PubMed
-
- Beal MF, et al. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321(6066):168–171. - PubMed
-
- Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293(5529):493–498. - PubMed
-
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19(5):233–238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
