

Electrostatics, structure prediction, and the energy landscapes for protein folding and binding
- PMID: 26183799
- PMCID: PMC4815325
- DOI: 10.1002/pro.2751
Electrostatics, structure prediction, and the energy landscapes for protein folding and binding
Abstract
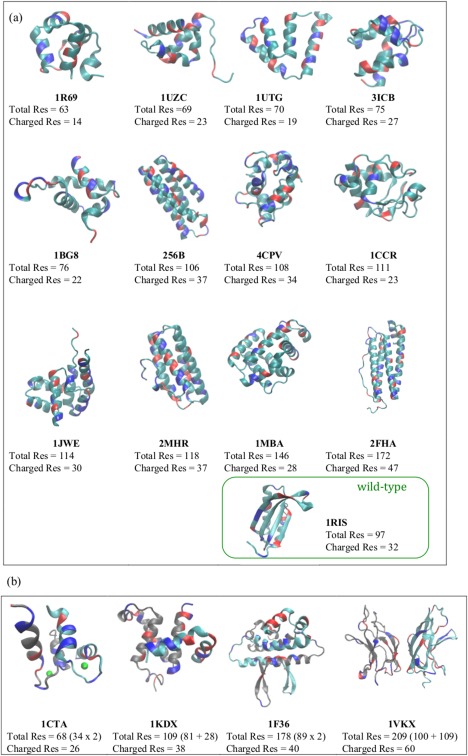
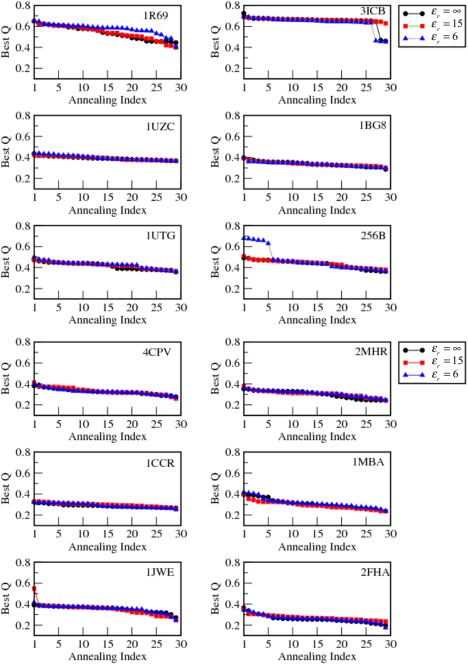
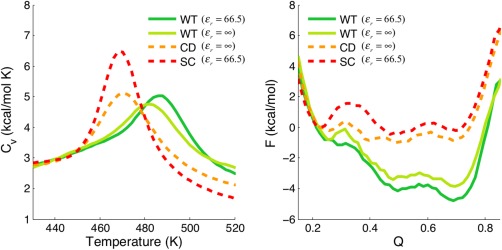
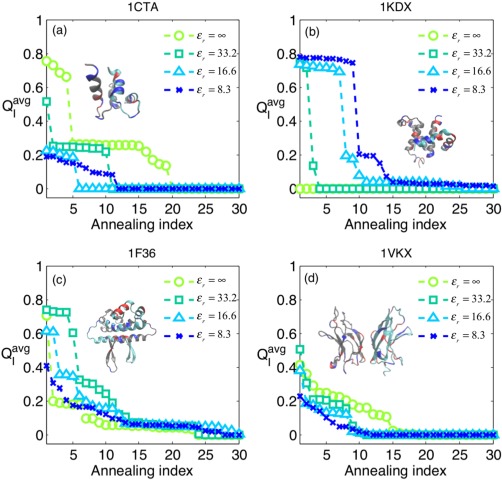
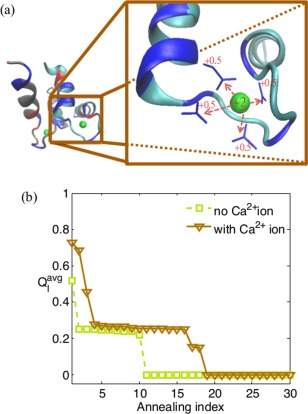
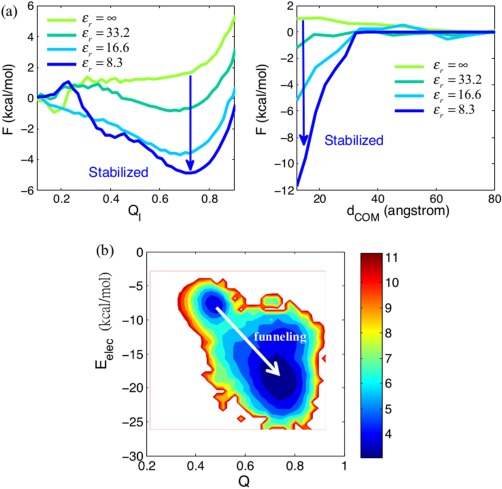
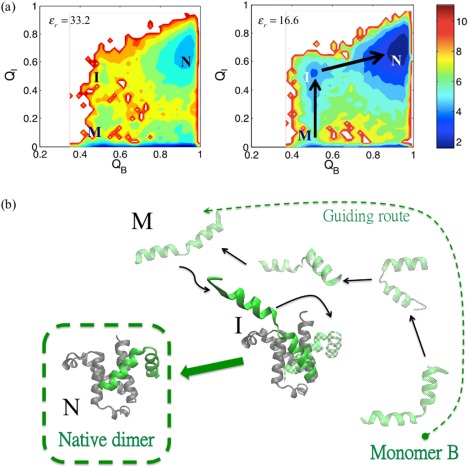
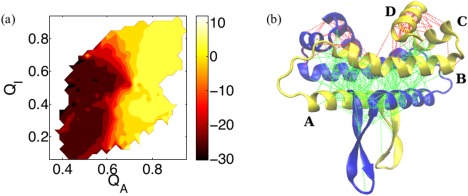
While being long in range and therefore weakly specific, electrostatic interactions are able to modulate the stability and folding landscapes of some proteins. The relevance of electrostatic forces for steering the docking of proteins to each other is widely acknowledged, however, the role of electrostatics in establishing specifically funneled landscapes and their relevance for protein structure prediction are still not clear. By introducing Debye-Hückel potentials that mimic long-range electrostatic forces into the Associative memory, Water mediated, Structure, and Energy Model (AWSEM), a transferable protein model capable of predicting tertiary structures, we assess the effects of electrostatics on the landscapes of thirteen monomeric proteins and four dimers. For the monomers, we find that adding electrostatic interactions does not improve structure prediction. Simulations of ribosomal protein S6 show, however, that folding stability depends monotonically on electrostatic strength. The trend in predicted melting temperatures of the S6 variants agrees with experimental observations. Electrostatic effects can play a range of roles in binding. The binding of the protein complex KIX-pKID is largely assisted by electrostatic interactions, which provide direct charge-charge stabilization of the native state and contribute to the funneling of the binding landscape. In contrast, for several other proteins, including the DNA-binding protein FIS, electrostatics causes frustration in the DNA-binding region, which favors its binding with DNA but not with its protein partner. This study highlights the importance of long-range electrostatics in functional responses to problems where proteins interact with their charged partners, such as DNA, RNA, as well as membranes.
Keywords: Debye-Hückel potentials; binding; electrostatically induced frustration; energy landscape theory; long-range electrostatics; protein folding; protein−protein interactions.
© 2015 The Protein Society.
Figures
Similar articles
-
Modulation of folding energy landscape by charge-charge interactions: linking experiments with computational modeling.Proc Natl Acad Sci U S A. 2015 Jan 20;112(3):E259-66. doi: 10.1073/pnas.1410424112. Epub 2015 Jan 6. Proc Natl Acad Sci U S A. 2015. PMID: 25564663 Free PMC article.
-
Electrostatics in protein binding and function.Curr Protein Pept Sci. 2002 Dec;3(6):601-14. doi: 10.2174/1389203023380431. Curr Protein Pept Sci. 2002. PMID: 12470214 Review.
-
Predictive energy landscapes for folding membrane protein assemblies.J Chem Phys. 2015 Dec 28;143(24):243101. doi: 10.1063/1.4929598. J Chem Phys. 2015. PMID: 26723586 Free PMC article.
-
The Role of Electrostatics and Folding Kinetics on the Thermostability of Homologous Cold Shock Proteins.J Chem Inf Model. 2020 Feb 24;60(2):546-561. doi: 10.1021/acs.jcim.9b00797. Epub 2020 Jan 17. J Chem Inf Model. 2020. PMID: 31910002 Free PMC article.
-
Protein electrostatics: a review of the equations and methods used to model electrostatic equations in biomolecules--applications in biotechnology.Biotechnol Annu Rev. 2003;9:315-95. doi: 10.1016/s1387-2656(03)09010-0. Biotechnol Annu Rev. 2003. PMID: 14650935 Review.
Cited by
-
Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics.Nucleic Acids Res. 2016 Jul 8;44(W1):W356-60. doi: 10.1093/nar/gkw304. Epub 2016 Apr 29. Nucleic Acids Res. 2016. PMID: 27131359 Free PMC article.
-
Smoothing a rugged protein folding landscape by sequence-based redesign.Sci Rep. 2016 Sep 26;6:33958. doi: 10.1038/srep33958. Sci Rep. 2016. PMID: 27667094 Free PMC article.
-
Modeling the Dynamics of Protein-Protein Interfaces, How and Why?Molecules. 2022 Mar 11;27(6):1841. doi: 10.3390/molecules27061841. Molecules. 2022. PMID: 35335203 Free PMC article. Review.
-
Features of molecular recognition of intrinsically disordered proteins via coupled folding and binding.Protein Sci. 2019 Nov;28(11):1952-1965. doi: 10.1002/pro.3718. Epub 2019 Sep 4. Protein Sci. 2019. PMID: 31441158 Free PMC article. Review.
-
Disorder Mediated Oligomerization of DISC1 Proteins Revealed by Coarse-Grained Molecular Dynamics Simulations.J Phys Chem B. 2019 Nov 14;123(45):9567-9575. doi: 10.1021/acs.jpcb.9b07467. Epub 2019 Oct 30. J Phys Chem B. 2019. PMID: 31614085 Free PMC article.
References
-
- Berg OG, von Hippel PH (1985) Diffusion‐controlled macromolecular interactions. Annu Rev Biophys Biophys Chem 14:131–160. - PubMed
-
- Honig B, Nicholls A (1995) Classical electrostatics in biology and chemistry. Science 268:1144–1149. - PubMed
-
- Schreiber G, Fersht AR (1996) Rapid, electrostatically assisted association of proteins. Nat Struct Biol 3:427–431. - PubMed
-
- Janin J (1997) The kinetics of protein‐protein recognition. Proteins 28:153–161. - PubMed
-
- Wolynes PG (2005) Energy landscapes and solved protein‐folding problems. Phil Trans Roy Soc A 363:453–464. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
