

Uridylation of RNA Hairpins by Tailor Confines the Emergence of MicroRNAs in Drosophila
- PMID: 26145176
- PMCID: PMC4518039
- DOI: 10.1016/j.molcel.2015.05.033
Uridylation of RNA Hairpins by Tailor Confines the Emergence of MicroRNAs in Drosophila
Abstract
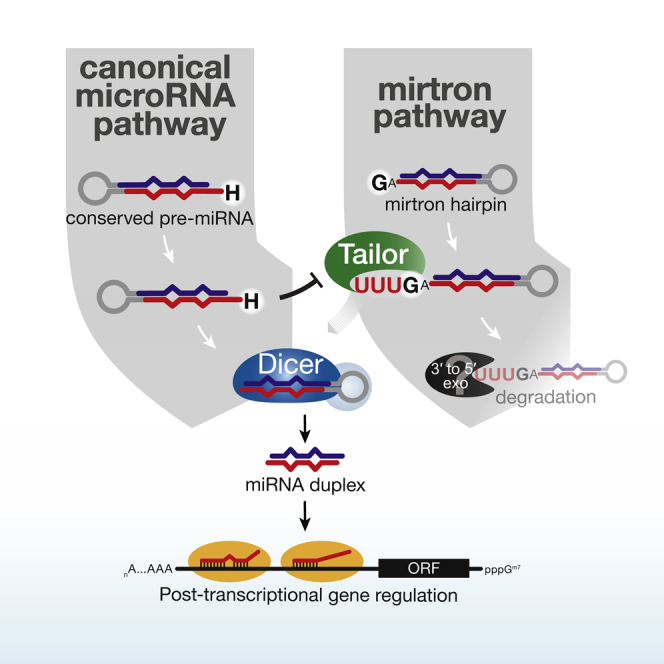
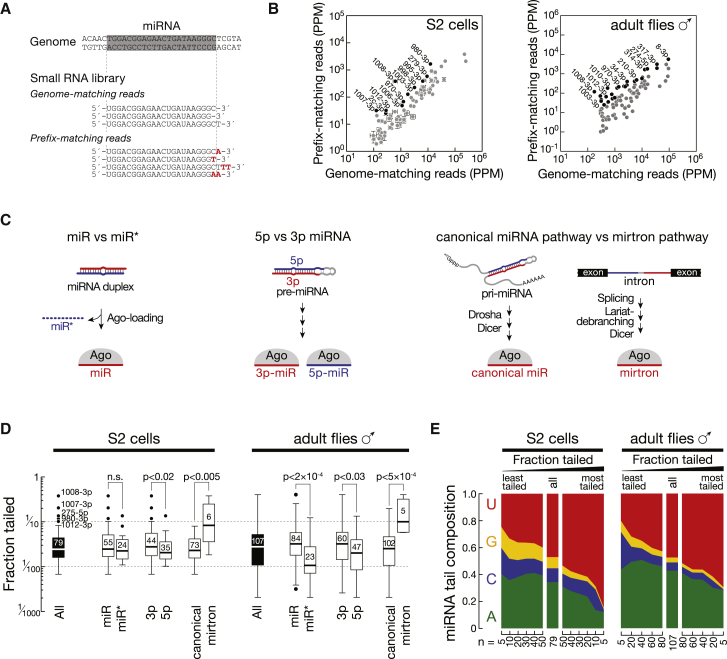
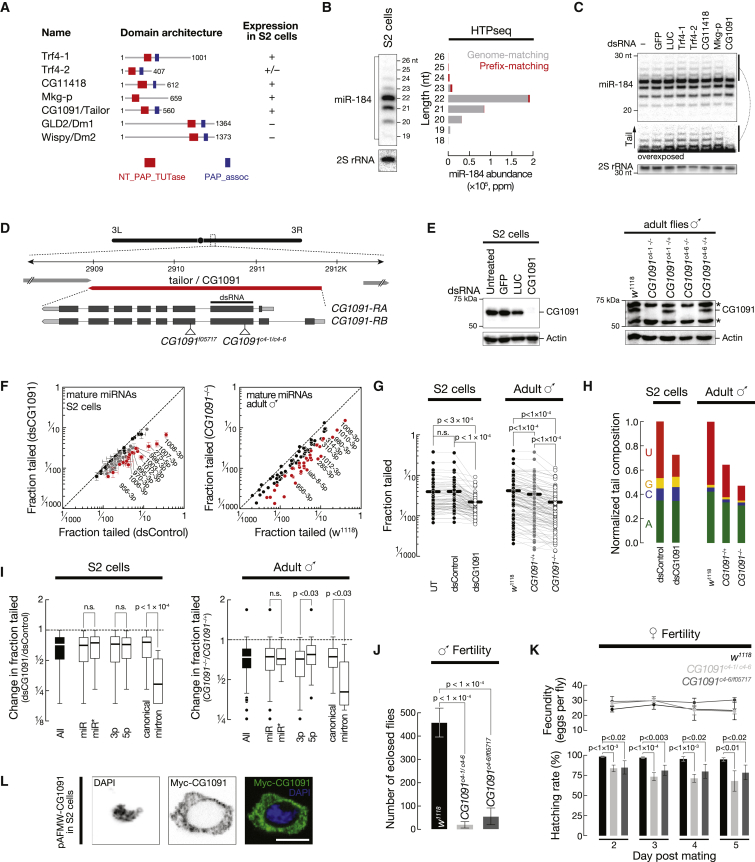
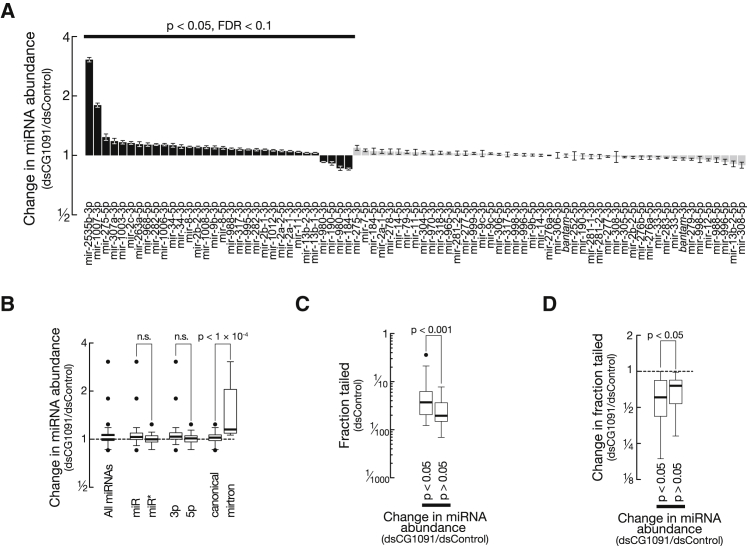
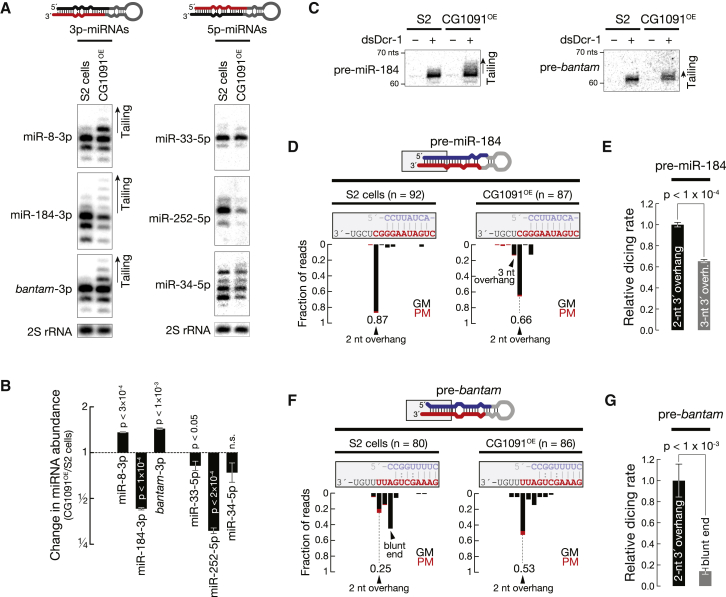
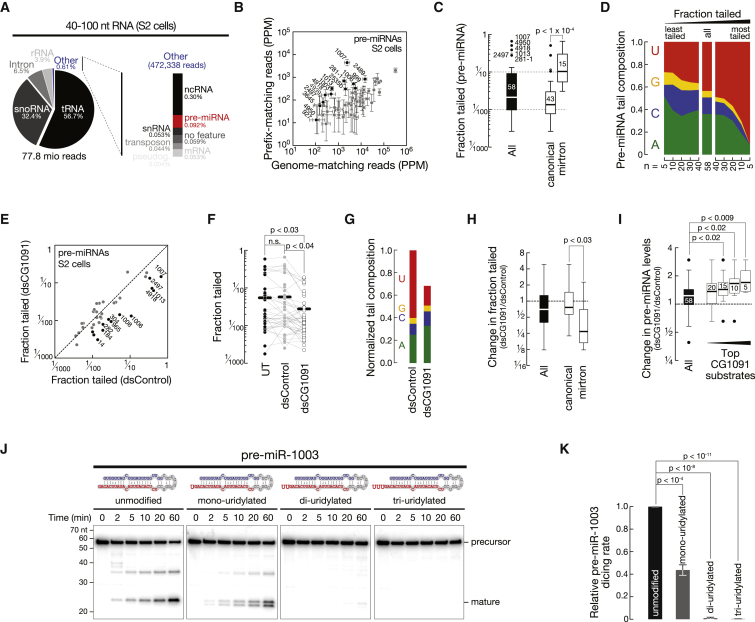
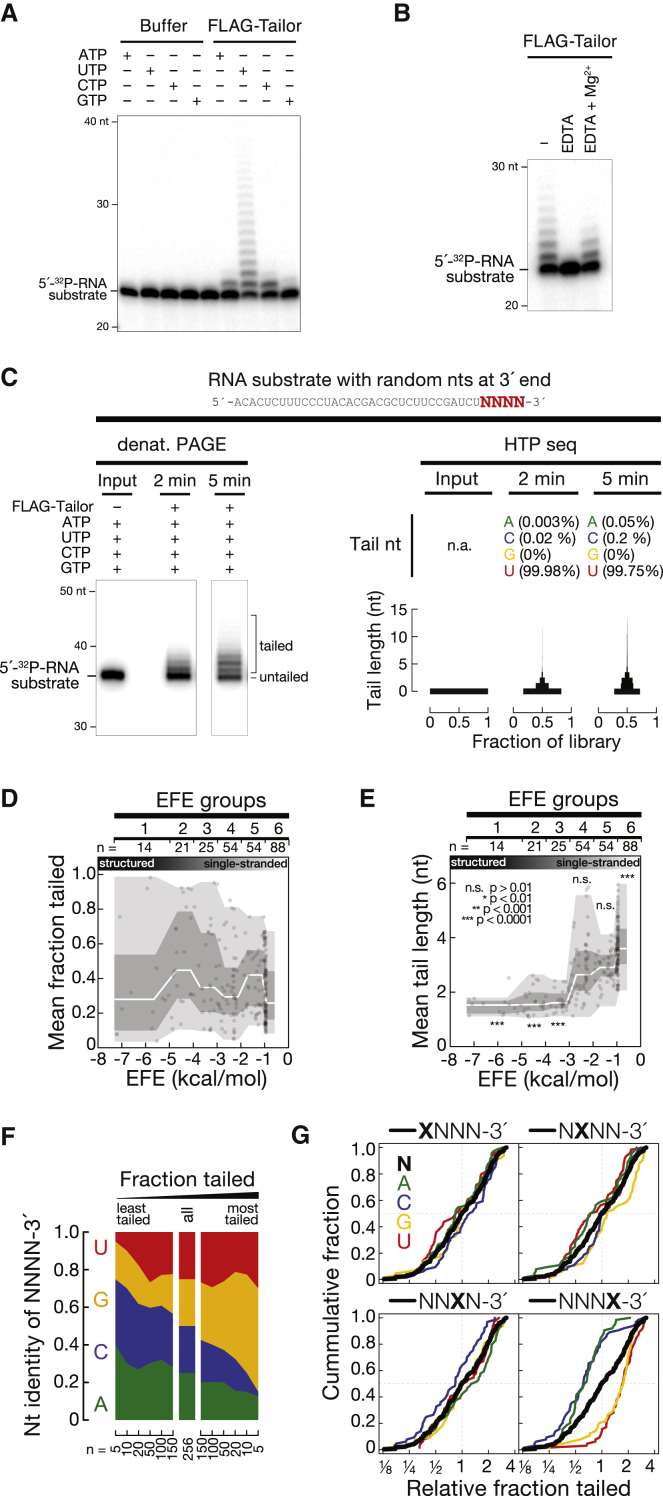
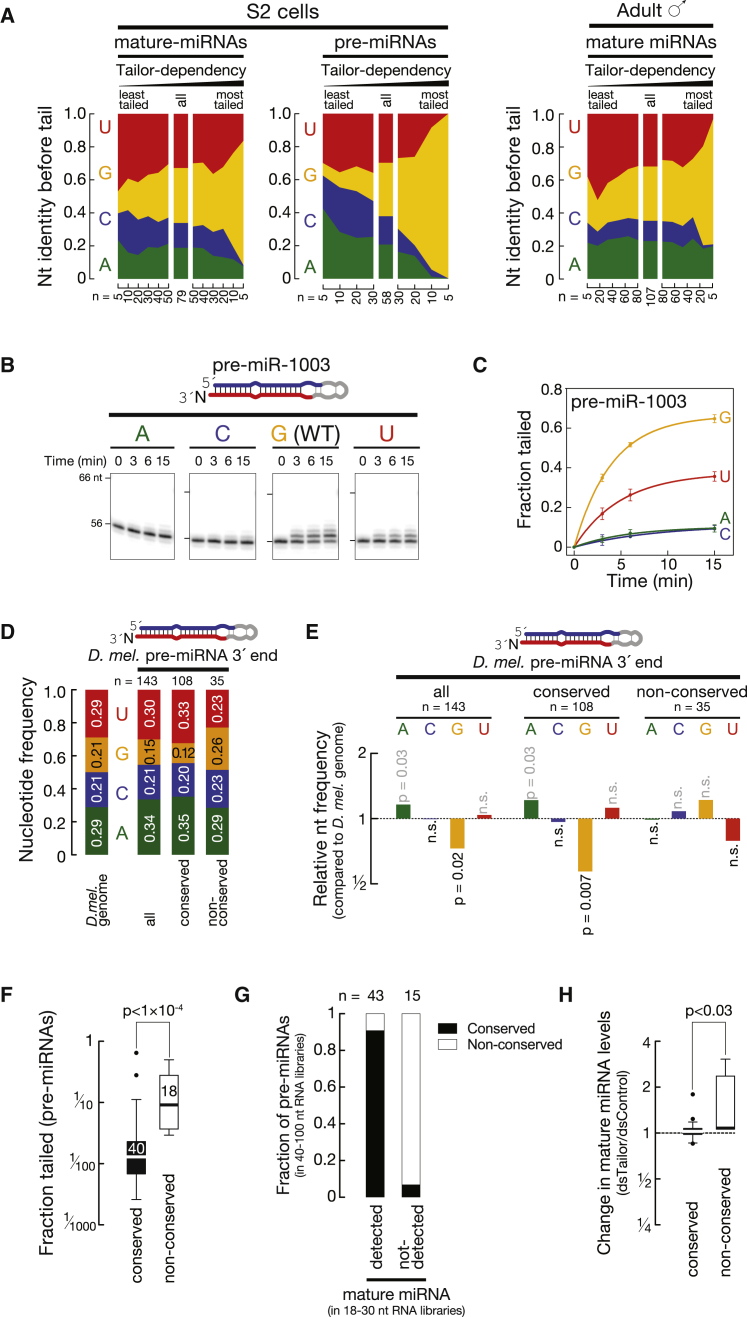
Uridylation of RNA species represents an emerging theme in post-transcriptional gene regulation. In the microRNA pathway, such modifications regulate small RNA biogenesis and stability in plants, worms, and mammals. Here, we report Tailor, an uridylyltransferase that is required for the majority of 3' end modifications of microRNAs in Drosophila and predominantly targets precursor hairpins. Uridylation modulates the characteristic two-nucleotide 3' overhang of microRNA hairpins, which regulates processing by Dicer-1 and destabilizes RNA hairpins. Tailor preferentially uridylates mirtron hairpins, thereby impeding the production of non-canonical microRNAs. Mirtron selectivity is explained by primary sequence specificity of Tailor, selecting substrates ending with a 3' guanosine. In contrast to mirtrons, conserved Drosophila precursor microRNAs are significantly depleted in 3' guanosine, thereby escaping regulatory uridylation. Our data support the hypothesis that evolutionary adaptation to Tailor-directed uridylation shapes the nucleotide composition of precursor microRNA 3' ends. Hence, hairpin uridylation may serve as a barrier for the de novo creation of microRNAs in Drosophila.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Selective Suppression of the Splicing-Mediated MicroRNA Pathway by the Terminal Uridyltransferase Tailor.Mol Cell. 2015 Jul 16;59(2):217-28. doi: 10.1016/j.molcel.2015.05.034. Epub 2015 Jul 2. Mol Cell. 2015. PMID: 26145174 Free PMC article.
-
The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila.Cell. 2007 Jul 13;130(1):89-100. doi: 10.1016/j.cell.2007.06.028. Epub 2007 Jun 28. Cell. 2007. PMID: 17599402 Free PMC article.
-
TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms.EMBO J. 2015 Jul 2;34(13):1801-15. doi: 10.15252/embj.201590931. Epub 2015 May 15. EMBO J. 2015. PMID: 25979828 Free PMC article.
-
Analysis of MicroRNA Function in Drosophila.Methods Mol Biol. 2016;1478:79-94. doi: 10.1007/978-1-4939-6371-3_4. Methods Mol Biol. 2016. PMID: 27730576 Review.
-
Gawky (GW) is the Drosophila melanogaster GW182 homologue.Adv Exp Med Biol. 2013;768:127-45. doi: 10.1007/978-1-4614-5107-5_8. Adv Exp Med Biol. 2013. PMID: 23224968 Review. No abstract available.
Cited by
-
AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2.Nat Commun. 2020 Jun 2;11(1):2765. doi: 10.1038/s41467-020-16533-w. Nat Commun. 2020. PMID: 32488030 Free PMC article.
-
Metazoan MicroRNAs.Cell. 2018 Mar 22;173(1):20-51. doi: 10.1016/j.cell.2018.03.006. Cell. 2018. PMID: 29570994 Free PMC article. Review.
-
Structure-function analysis of microRNA 3'-end trimming by Nibbler.Proc Natl Acad Sci U S A. 2020 Dec 1;117(48):30370-30379. doi: 10.1073/pnas.2018156117. Epub 2020 Nov 16. Proc Natl Acad Sci U S A. 2020. PMID: 33199607 Free PMC article.
-
TENT2, TUT4, and TUT7 selectively regulate miRNA sequence and abundance.Nat Commun. 2022 Sep 7;13(1):5260. doi: 10.1038/s41467-022-32969-8. Nat Commun. 2022. PMID: 36071058 Free PMC article.
-
Prevalent cytidylation and uridylation of precursor miRNAs in Arabidopsis.Nat Plants. 2019 Dec;5(12):1260-1272. doi: 10.1038/s41477-019-0562-1. Epub 2019 Dec 2. Nat Plants. 2019. PMID: 31792392
References
-
- Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. - PubMed
-
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
