

Fulfilling the Promise of "Biased" G Protein-Coupled Receptor Agonism
- PMID: 26134495
- PMCID: PMC4551052
- DOI: 10.1124/mol.115.099630
Fulfilling the Promise of "Biased" G Protein-Coupled Receptor Agonism
Abstract
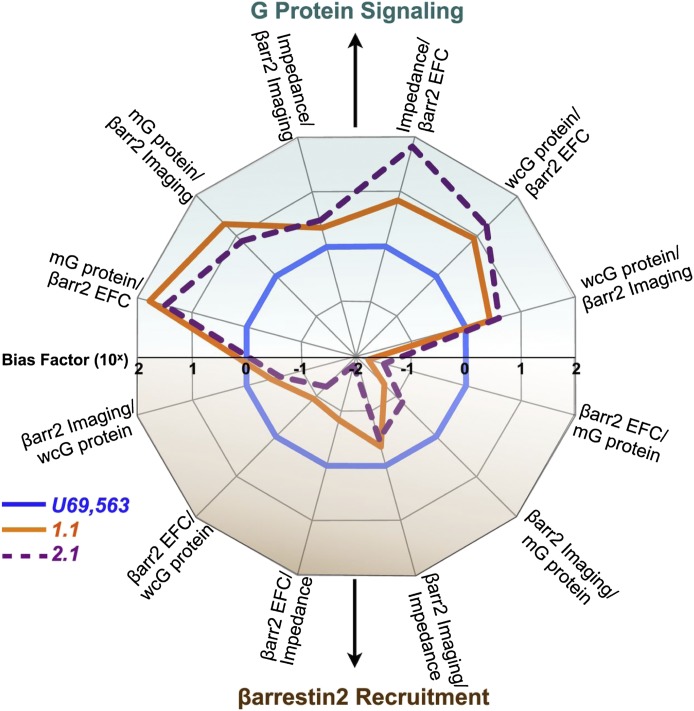
The fact that over 30% of current pharmaceuticals target heptahelical G protein-coupled receptors (GPCRs) attests to their tractability as drug targets. Although GPCR drug development has traditionally focused on conventional agonists and antagonists, the growing appreciation that GPCRs mediate physiologically relevant effects via both G protein and non-G protein effectors has prompted the search for ligands that can "bias" downstream signaling in favor of one or the other process. Biased ligands are novel entities with distinct signaling profiles dictated by ligand structure, and the potential prospect of biased ligands as better drugs has been pleonastically proclaimed. Indeed, preclinical proof-of-concept studies have demonstrated that both G protein and arrestin pathway-selective ligands can promote beneficial effects in vivo while simultaneously antagonizing deleterious ones. But along with opportunity comes added complexity and new challenges for drug discovery. If ligands can be biased, then ligand classification becomes assay dependent, and more nuanced screening approaches are needed to capture ligand efficacy across several dimensions of signaling. Moreover, because the signaling repertoire of biased ligands differs from that of the native agonist, unpredicted responses may arise in vivo as these unbalanced signals propagate. For any given GPCR target, establishing a framework relating in vitro efficacy to in vivo biologic response is crucial to biased drug discovery. This review discusses approaches to describing ligand efficacy in vitro, translating ligand bias into biologic response, and developing a systems-level understanding of biased agonism in vivo, with the overall goal of overcoming current barriers to developing biased GPCR therapeutics.
Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics.
Figures

Similar articles
-
Translating in vitro ligand bias into in vivo efficacy.Cell Signal. 2018 Jan;41:46-55. doi: 10.1016/j.cellsig.2017.05.002. Epub 2017 May 7. Cell Signal. 2018. PMID: 28495495 Free PMC article. Review.
-
Arrestin pathways as drug targets.Prog Mol Biol Transl Sci. 2013;118:469-97. doi: 10.1016/B978-0-12-394440-5.00018-8. Prog Mol Biol Transl Sci. 2013. PMID: 23764065 Review.
-
Biased agonism at G protein-coupled receptors: the promise and the challenges--a medicinal chemistry perspective.Med Res Rev. 2014 Nov;34(6):1286-330. doi: 10.1002/med.21318. Epub 2014 May 5. Med Res Rev. 2014. PMID: 24796277 Review.
-
Beta-arrestin biased agonism/antagonism at cardiovascular seven transmembrane-spanning receptors.Curr Pharm Des. 2012;18(2):192-8. doi: 10.2174/138161212799040475. Curr Pharm Des. 2012. PMID: 22229558 Review.
-
Minireview: More than just a hammer: ligand "bias" and pharmaceutical discovery.Mol Endocrinol. 2014 Mar;28(3):281-94. doi: 10.1210/me.2013-1314. Epub 2014 Jan 16. Mol Endocrinol. 2014. PMID: 24433041 Free PMC article. Review.
Cited by
-
Biased signaling in naturally occurring mutations of G protein-coupled receptors associated with diverse human diseases.Biochim Biophys Acta Mol Basis Dis. 2021 Jan 1;1867(1):165973. doi: 10.1016/j.bbadis.2020.165973. Epub 2020 Sep 17. Biochim Biophys Acta Mol Basis Dis. 2021. PMID: 32949766 Free PMC article. Review.
-
High-throughput screening against protein:protein interaction interfaces reveals anti-cancer therapeutics as potent modulators of the voltage-gated Na+ channel complex.Sci Rep. 2019 Nov 15;9(1):16890. doi: 10.1038/s41598-019-53110-8. Sci Rep. 2019. PMID: 31729429 Free PMC article.
-
Comparison of the pharmacological profiles of arginine vasopressin and oxytocin analogs at marmoset, macaque, and human vasopressin 1a receptor.Biomed Pharmacother. 2020 Jun;126:110060. doi: 10.1016/j.biopha.2020.110060. Epub 2020 Mar 4. Biomed Pharmacother. 2020. PMID: 32145592 Free PMC article.
-
GIT2 Acts as a Systems-Level Coordinator of Neurometabolic Activity and Pathophysiological Aging.Front Endocrinol (Lausanne). 2016 Jan 18;6:191. doi: 10.3389/fendo.2015.00191. eCollection 2015. Front Endocrinol (Lausanne). 2016. PMID: 26834700 Free PMC article.
-
Female-specific myoinhibitory peptide neurons regulate mating receptivity in Drosophila melanogaster.Nat Commun. 2017 Nov 21;8(1):1630. doi: 10.1038/s41467-017-01794-9. Nat Commun. 2017. PMID: 29158481 Free PMC article.
References
-
- Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjølbye AL, Sheikh SP, Hansen JL. (2007) Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin Pharmacol Toxicol 100:296–301. - PubMed
-
- Appleton KM, Luttrell LM. (2013) Emergent biological properties of arrestin pathway-selective biased agonism. J Recept Signal Transduct Res 33:153–161. - PubMed
-
- Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ. (2000) Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat Immunol 1:227–233. - PubMed
-
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122:261–273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01-GM095497/GM/NIGMS NIH HHS/United States
- R01 GM095497/GM/NIGMS NIH HHS/United States
- R01 DK055524/DK/NIDDK NIH HHS/United States
- R01 DA033073/DA/NIDA NIH HHS/United States
- R01 DA031927/DA/NIDA NIH HHS/United States
- P01-DA009158/DA/NIDA NIH HHS/United States
- P01 DA009158/DA/NIDA NIH HHS/United States
- R01-DA033073/DA/NIDA NIH HHS/United States
- R01-DA038964/DA/NIDA NIH HHS/United States
- R01-DK055524/DK/NIDDK NIH HHS/United States
- R01 DA038964/DA/NIDA NIH HHS/United States
- R01-DA031927/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources