

Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice
- PMID: 26124132
- PMCID: PMC4522794
- DOI: 10.1073/pnas.1505819112
Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice
Abstract
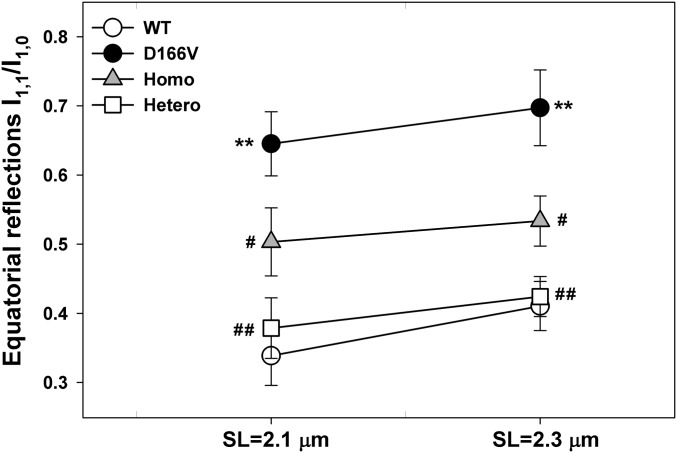
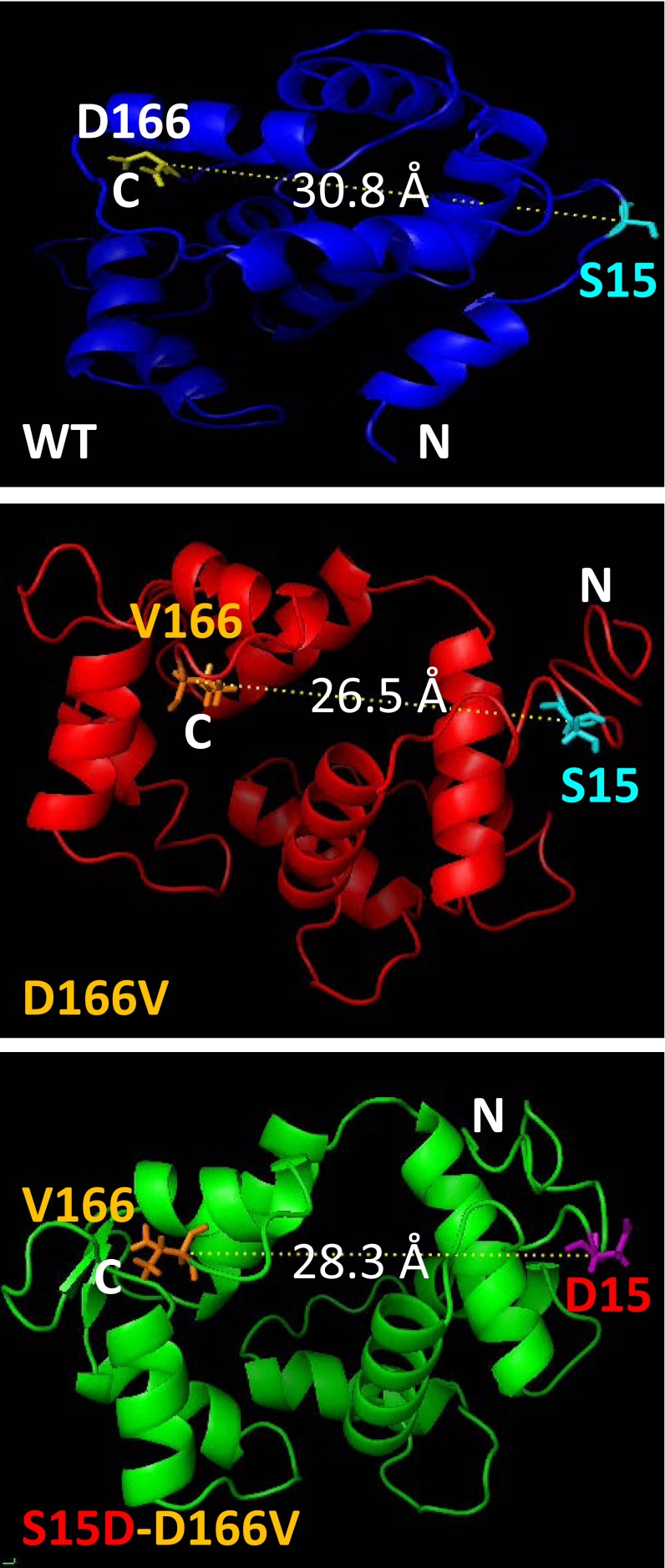
Myosin light chain kinase (MLCK)-dependent phosphorylation of the regulatory light chain (RLC) of cardiac myosin is known to play a beneficial role in heart disease, but the idea of a phosphorylation-mediated reversal of a hypertrophic cardiomyopathy (HCM) phenotype is novel. Our previous studies on transgenic (Tg) HCM-RLC mice revealed that the D166V (Aspartate166 → Valine) mutation-induced changes in heart morphology and function coincided with largely reduced RLC phosphorylation in situ. We hypothesized that the introduction of a constitutively phosphorylated Serine15 (S15D) into the hearts of D166V mice would prevent the development of a deleterious HCM phenotype. In support of this notion, MLCK-induced phosphorylation of D166V-mutated hearts was found to rescue some of their abnormal contractile properties. Tg-S15D-D166V mice were generated with the human cardiac RLC-S15D-D166V construct substituted for mouse cardiac RLC and were subjected to functional, structural, and morphological assessments. The results were compared with Tg-WT and Tg-D166V mice expressing the human ventricular RLC-WT or its D166V mutant, respectively. Echocardiography and invasive hemodynamic studies demonstrated significant improvements of intact heart function in S15D-D166V mice compared with D166V, with the systolic and diastolic indices reaching those monitored in WT mice. A largely reduced maximal tension and abnormally high myofilament Ca(2+) sensitivity observed in D166V-mutated hearts were reversed in S15D-D166V mice. Low-angle X-ray diffraction study revealed that altered myofilament structures present in HCM-D166V mice were mitigated in S15D-D166V rescue mice. Our collective results suggest that expression of pseudophosphorylated RLC in the hearts of HCM mice is sufficient to prevent the development of the pathological HCM phenotype.
Keywords: X-ray structure; cardiomyopathy; hemodynamics; myocardial contraction; myosin RLC.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Myosin light chain phosphorylation to the rescue.Proc Natl Acad Sci U S A. 2015 Jul 28;112(30):9148-9. doi: 10.1073/pnas.1511455112. Epub 2015 Jul 8. Proc Natl Acad Sci U S A. 2015. PMID: 26157138 Free PMC article. No abstract available.
Similar articles
-
Therapeutic potential of AAV9-S15D-RLC gene delivery in humanized MYL2 mouse model of HCM.J Mol Med (Berl). 2019 Jul;97(7):1033-1047. doi: 10.1007/s00109-019-01791-z. Epub 2019 May 17. J Mol Med (Berl). 2019. PMID: 31101927 Free PMC article.
-
Mechanistic basis for rescuing hypertrophic cardiomyopathy with myosin regulatory light chain phosphorylation.Cytoskeleton (Hoboken). 2024 Dec;81(12):806-814. doi: 10.1002/cm.21854. Epub 2024 Mar 17. Cytoskeleton (Hoboken). 2024. PMID: 38494592 Free PMC article.
-
In vitro rescue study of a malignant familial hypertrophic cardiomyopathy phenotype by pseudo-phosphorylation of myosin regulatory light chain.Arch Biochem Biophys. 2014 Jun 15;552-553:29-39. doi: 10.1016/j.abb.2013.12.011. Epub 2013 Dec 26. Arch Biochem Biophys. 2014. PMID: 24374283 Free PMC article.
-
Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations.J Muscle Res Cell Motil. 2015 Dec;36(6):433-45. doi: 10.1007/s10974-015-9423-3. Epub 2015 Sep 18. J Muscle Res Cell Motil. 2015. PMID: 26385864 Free PMC article. Review.
-
Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains.Pflugers Arch. 2019 May;471(5):683-699. doi: 10.1007/s00424-019-02257-4. Epub 2019 Jan 31. Pflugers Arch. 2019. PMID: 30706179 Free PMC article. Review.
Cited by
-
Revisiting Frank-Starling: regulatory light chain phosphorylation alters the rate of force redevelopment (ktr ) in a length-dependent fashion.J Physiol. 2016 Sep 15;594(18):5237-54. doi: 10.1113/JP272441. Epub 2016 Jul 24. J Physiol. 2016. PMID: 27291932 Free PMC article.
-
Myofilament dysfunction in diastolic heart failure.Heart Fail Rev. 2024 Jan;29(1):79-93. doi: 10.1007/s10741-023-10352-z. Epub 2023 Oct 14. Heart Fail Rev. 2024. PMID: 37837495 Free PMC article. Review.
-
Integrated Quantitative Phosphoproteomics and Cell-based Functional Screening Reveals Specific Pathological Cardiac Hypertrophy-related Phosphorylation Sites.Mol Cells. 2021 Jul 31;44(7):500-516. doi: 10.14348/molcells.2021.4002. Mol Cells. 2021. PMID: 34158421 Free PMC article.
-
Distinct sequences and post-translational modifications in cardiac atrial and ventricular myosin light chains revealed by top-down mass spectrometry.J Mol Cell Cardiol. 2017 Jun;107:13-21. doi: 10.1016/j.yjmcc.2017.04.002. Epub 2017 Apr 17. J Mol Cell Cardiol. 2017. PMID: 28427997 Free PMC article.
-
Cardiac Sarcomere Signaling in Health and Disease.Int J Mol Sci. 2022 Dec 19;23(24):16223. doi: 10.3390/ijms232416223. Int J Mol Sci. 2022. PMID: 36555864 Free PMC article. Review.
References
-
- Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J Cardiovasc Electrophysiol. 2008;19(1):104–110. - PubMed
-
- Richard P, et al. EUROGENE Heart Failure Project 2003. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107(17):2227–2232, and erratum (2004) 109(25):3258.
-
- Kerrick WG, Kazmierczak K, Xu Y, Wang Y, Szczesna-Cordary D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009;23(3):855–865. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
