

ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer
- PMID: 26122181
- PMCID: PMC4911210
- DOI: 10.1038/nrclinonc.2015.117
ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer
Abstract
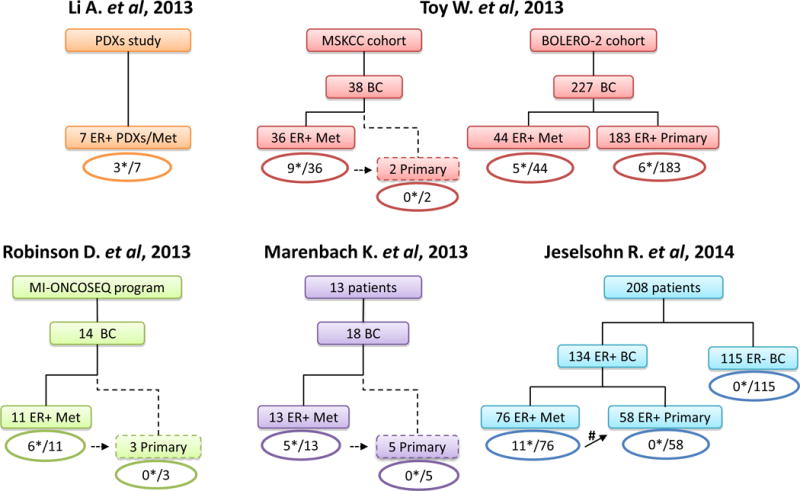
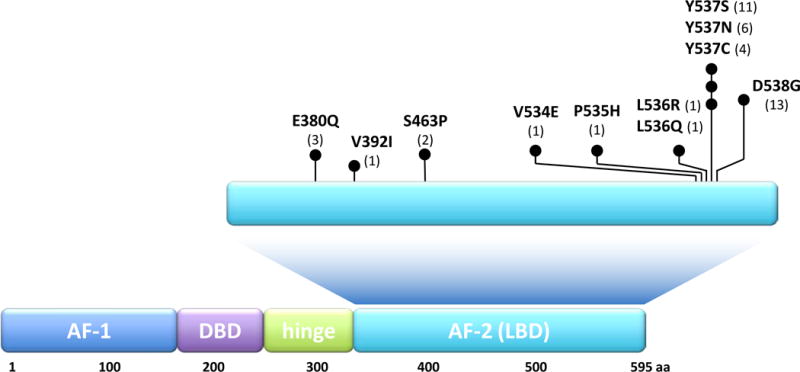
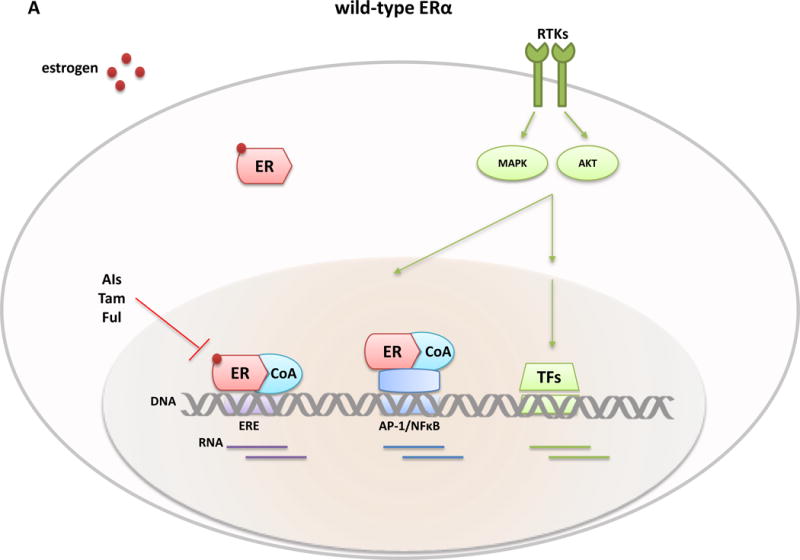
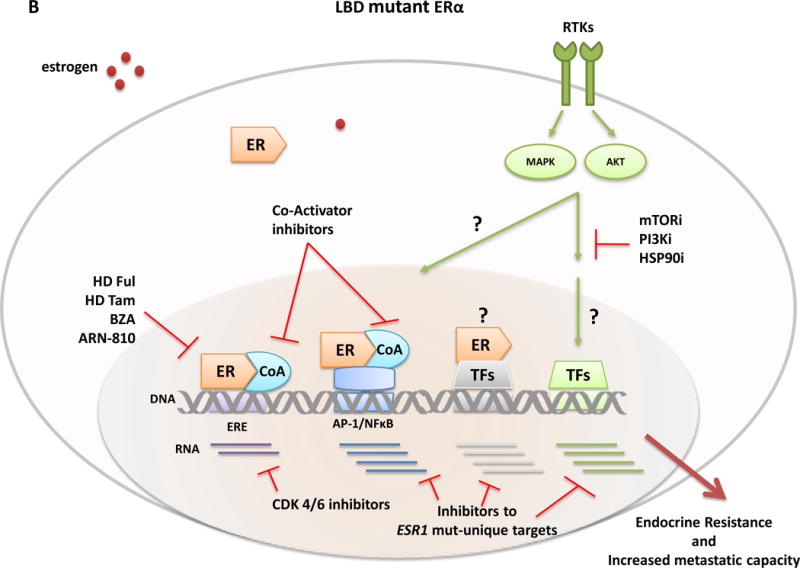
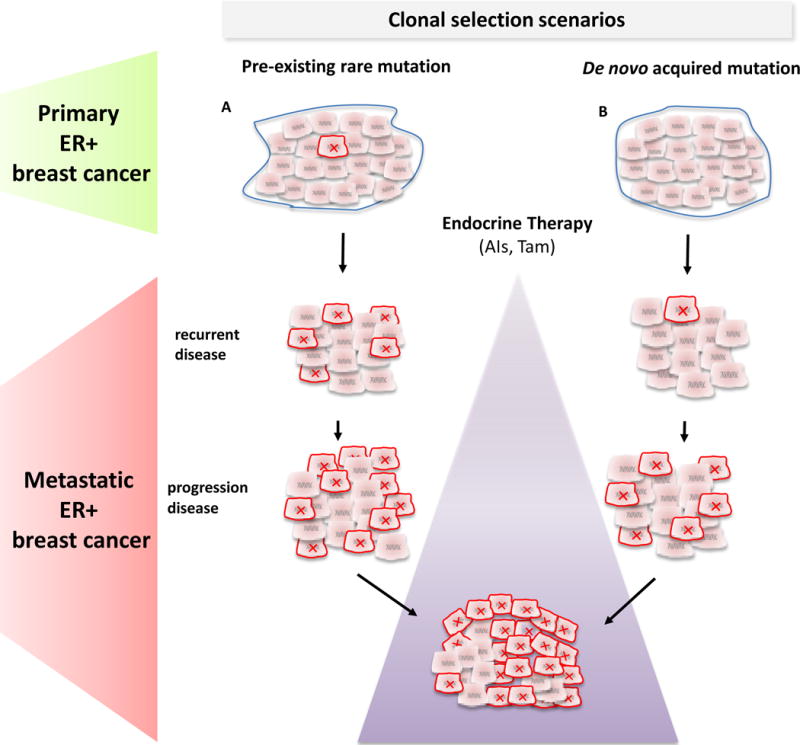
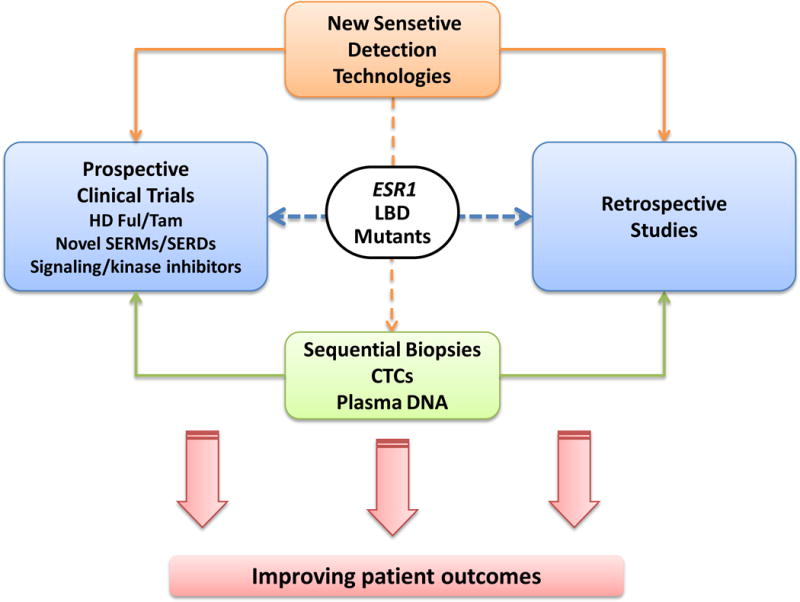
Approximately 70% of breast cancers are oestrogen receptor α (ER) positive, and are, therefore, treated with endocrine therapies. However, about 25% of patients with primary disease and almost all patients with metastases will present with or eventually develop endocrine resistance. Despite the magnitude of this clinical challenge, the mechanisms underlying the development of resistance remain largely unknown. In the past 2 years, several studies unveiled gain-of-function mutations in ESR1, the gene encoding the ER, in approximately 20% of patients with metastatic ER-positive disease who received endocrine therapies, such as tamoxifen and aromatase inhibitors. These mutations are clustered in a 'hotspot' within the ligand-binding domain (LBD) of the ER and lead to ligand-independent ER activity that promotes tumour growth, partial resistance to endocrine therapy, and potentially enhanced metastatic capacity; thus, ER LBD mutations might account for a mechanism of acquired endocrine resistance in a substantial fraction of patients with metastatic disease. In general, the absence of detectable ESR1 mutations in patients with treatment-naive disease, and the correlation between the frequency of patients with tumours harbouring these mutations and the number of endocrine treatments received suggest that, under selective treatment pressure, clonal expansion of rare mutant clones occurs, leading to resistance. Preclinical and clinical development of rationale-based novel therapeutic strategies that inhibit these ER mutants has the potential to substantially improve treatment outcomes. We discuss the contribution of ESR1 mutations to the development of acquired resistance to endocrine therapy, and evaluate how mutated ER can be detected and targeted to overcome resistance and improve patient outcomes.
Conflict of interest statement
RS has received research funding from AstraZenca and GlaxoSmithKline. MB has received a commercial research grant and is a consultant/advisory board member for Novartis Pharmaceuticals.
Figures
Similar articles
-
The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials.Biochim Biophys Acta Rev Cancer. 2019 Dec;1872(2):188315. doi: 10.1016/j.bbcan.2019.188315. Epub 2019 Oct 21. Biochim Biophys Acta Rev Cancer. 2019. PMID: 31647985 Review.
-
The prevalence of estrogen receptor-1 mutation in advanced breast cancer: The estrogen receptor one study (EROS1).Cancer Treat Res Commun. 2019;19:100123. doi: 10.1016/j.ctarc.2019.100123. Epub 2019 Feb 21. Cancer Treat Res Commun. 2019. PMID: 30826563
-
Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer.Ann Oncol. 2018 Apr 1;29(4):872-880. doi: 10.1093/annonc/mdy025. Ann Oncol. 2018. PMID: 29360925 Free PMC article.
-
Activating ESR1 mutations in hormone-resistant metastatic breast cancer.Nat Genet. 2013 Dec;45(12):1446-51. doi: 10.1038/ng.2823. Epub 2013 Nov 3. Nat Genet. 2013. PMID: 24185510 Free PMC article.
-
The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer.Curr Oncol Rep. 2017 May;19(5):35. doi: 10.1007/s11912-017-0591-8. Curr Oncol Rep. 2017. PMID: 28374222 Review.
Cited by
-
ESR1 Gene Mutation in Hormone Receptor-Positive HER2-Negative Metastatic Breast Cancer Patients: Concordance Between Tumor Tissue and Circulating Tumor DNA Analysis.Front Oncol. 2021 Mar 11;11:625636. doi: 10.3389/fonc.2021.625636. eCollection 2021. Front Oncol. 2021. PMID: 33777770 Free PMC article.
-
Point-activated ESR1Y541S has a dramatic effect on the development of sexually dimorphic organs.Genes Dev. 2020 Oct 1;34(19-20):1304-1309. doi: 10.1101/gad.339424.120. Epub 2020 Sep 10. Genes Dev. 2020. PMID: 32912899 Free PMC article.
-
Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer.Nat Rev Clin Oncol. 2024 Oct;21(10):743-761. doi: 10.1038/s41571-024-00935-6. Epub 2024 Aug 23. Nat Rev Clin Oncol. 2024. PMID: 39179659 Review.
-
Rare subclonal sequencing of breast cancers indicates putative metastatic driver mutations are predominately acquired after dissemination.Genome Med. 2024 Feb 6;16(1):26. doi: 10.1186/s13073-024-01293-9. Genome Med. 2024. PMID: 38321573 Free PMC article.
-
The Emerging Role of Extracellular Vesicles in Endocrine Resistant Breast Cancer.Cancers (Basel). 2021 Mar 8;13(5):1160. doi: 10.3390/cancers13051160. Cancers (Basel). 2021. PMID: 33800302 Free PMC article. Review.
References
-
- Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
