

Myosin regulatory light chain phosphorylation enhances cardiac β-myosin in vitro motility under load
- PMID: 26116789
- PMCID: PMC4790447
- DOI: 10.1016/j.abb.2015.06.014
Myosin regulatory light chain phosphorylation enhances cardiac β-myosin in vitro motility under load
Abstract
Familial hypertrophic cardiomyopathy (HCM) is characterized by left ventricular hypertrophy and myofibrillar disarray, and often results in sudden cardiac death. Two HCM mutations, N47K and R58Q, are located in the myosin regulatory light chain (RLC). The RLC mechanically stabilizes the myosin lever arm, which is crucial to myosin's ability to transmit contractile force. The N47K and R58Q mutations have previously been shown to reduce actin filament velocity under load, stemming from a more compliant lever arm (Greenberg, 2010). In contrast, RLC phosphorylation was shown to impart stiffness to the myosin lever arm (Greenberg, 2009). We hypothesized that phosphorylation of the mutant HCM-RLC may mitigate distinct mutation-induced structural and functional abnormalities. In vitro motility assays were utilized to investigate the effects of RLC phosphorylation on the HCM-RLC mutant phenotype in the presence of an α-actinin frictional load. Porcine cardiac β-myosin was depleted of its native RLC and reconstituted with mutant or wild-type human RLC in phosphorylated or non-phosphorylated form. Consistent with previous findings, in the presence of load, myosin bearing the HCM mutations reduced actin sliding velocity compared to WT resulting in 31-41% reductions in force production. Myosin containing phosphorylated RLC (WT or mutant) increased sliding velocity and also restored mutant myosin force production to near WT unphosphorylated values. These results point to RLC phosphorylation as a general mechanism to increase force production of the individual myosin motor and as a potential target to ameliorate the HCM-induced phenotype at the molecular level.
Keywords: Cardiac ventricular myosin; Hypertrophic cardiomyopathy; Load dependence; Regulatory light chain phosphorylation.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
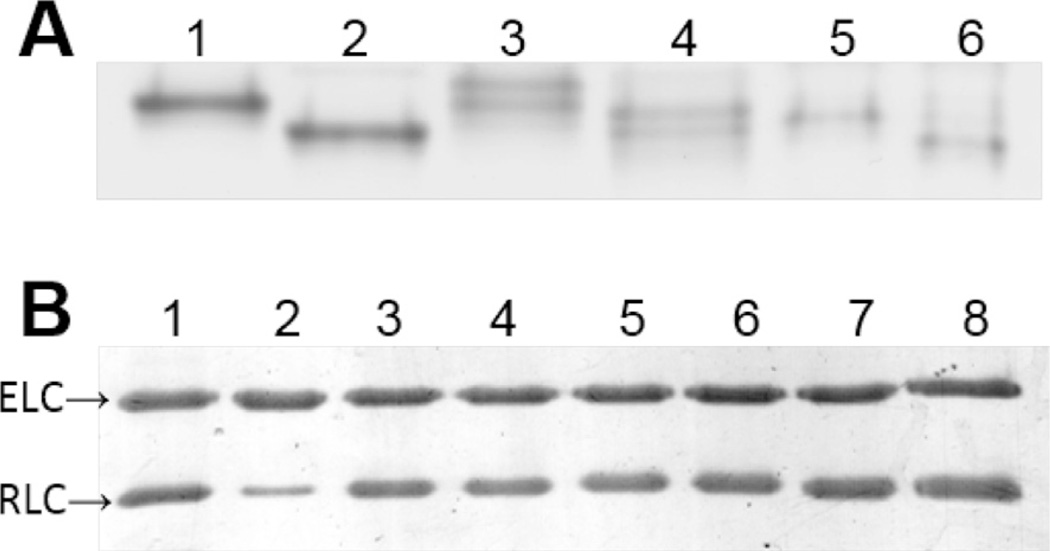
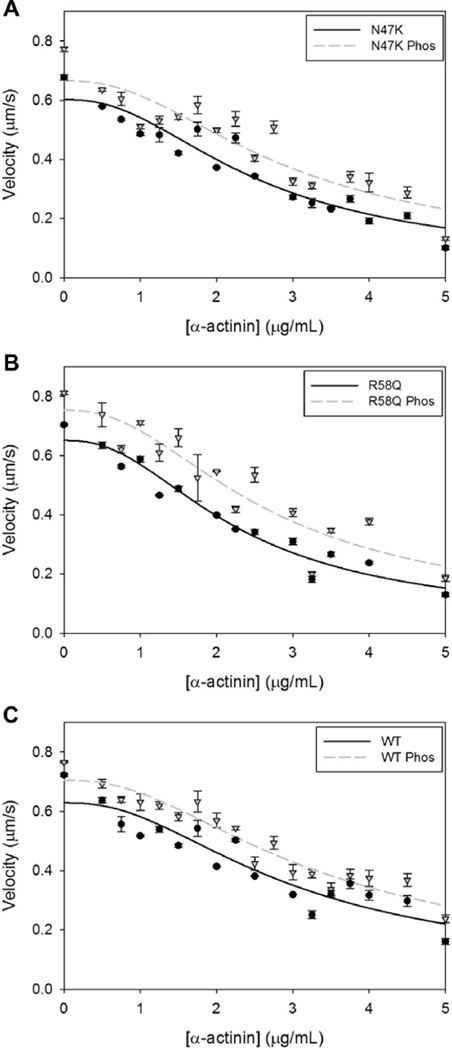

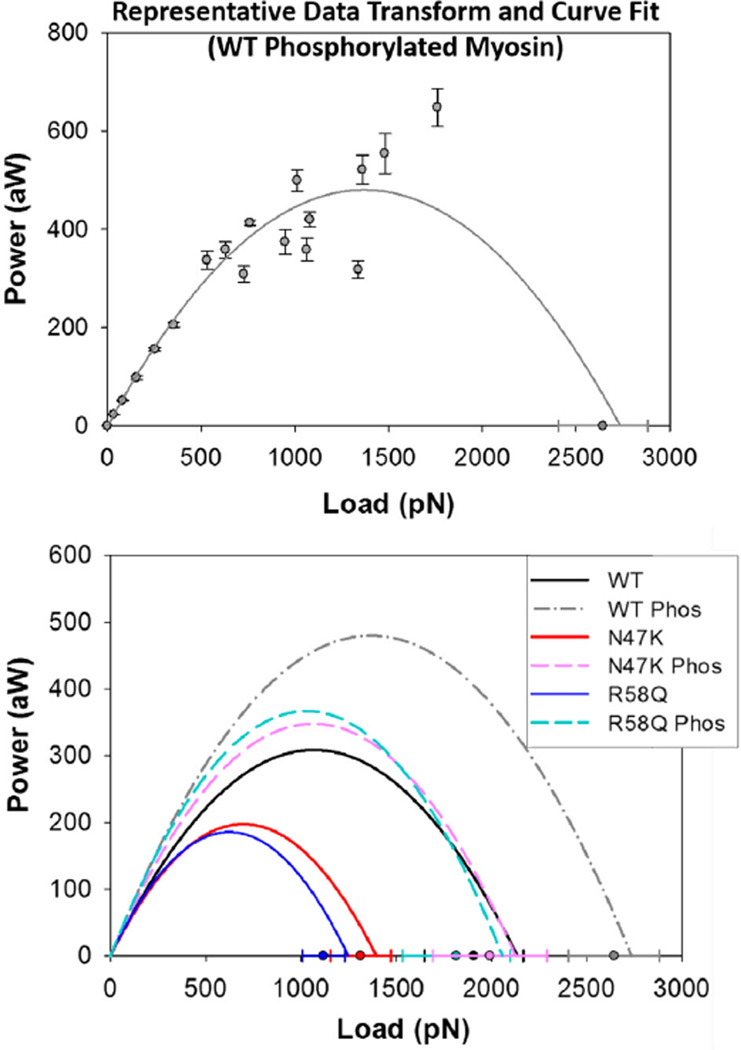
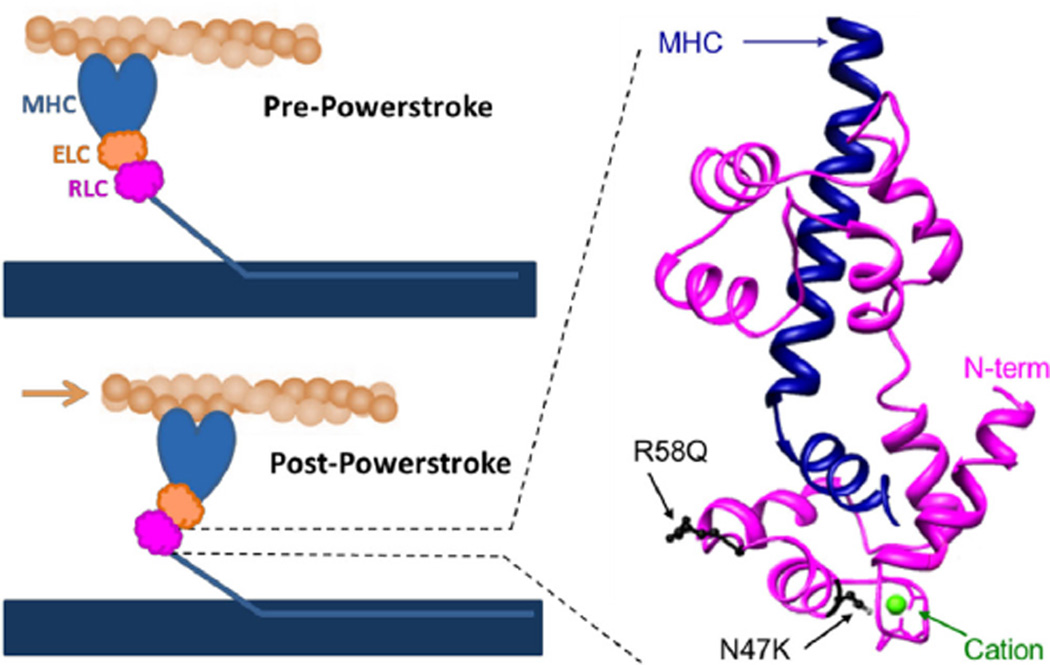
Similar articles
-
Impact of familial hypertrophic cardiomyopathy-linked mutations in the NH2 terminus of the RLC on β-myosin cross-bridge mechanics.J Appl Physiol (1985). 2014 Dec 15;117(12):1471-7. doi: 10.1152/japplphysiol.00798.2014. Epub 2014 Oct 16. J Appl Physiol (1985). 2014. PMID: 25324513 Free PMC article.
-
Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain.FEBS J. 2019 Jan;286(1):151-168. doi: 10.1111/febs.14702. Epub 2018 Dec 1. FEBS J. 2019. PMID: 30430732 Free PMC article.
-
Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics.J Mol Cell Cardiol. 2009 Jan;46(1):108-15. doi: 10.1016/j.yjmcc.2008.09.126. Epub 2008 Sep 27. J Mol Cell Cardiol. 2009. PMID: 18929571 Free PMC article.
-
Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations.J Muscle Res Cell Motil. 2015 Dec;36(6):433-45. doi: 10.1007/s10974-015-9423-3. Epub 2015 Sep 18. J Muscle Res Cell Motil. 2015. PMID: 26385864 Free PMC article. Review.
-
Regulatory light chains of striated muscle myosin. Structure, function and malfunction.Curr Drug Targets Cardiovasc Haematol Disord. 2003 Jun;3(2):187-97. doi: 10.2174/1568006033481474. Curr Drug Targets Cardiovasc Haematol Disord. 2003. PMID: 12769642 Review.
Cited by
-
Gene expression patterns in transgenic mouse models of hypertrophic cardiomyopathy caused by mutations in myosin regulatory light chain.Arch Biochem Biophys. 2016 Jul 1;601:121-32. doi: 10.1016/j.abb.2016.02.022. Epub 2016 Feb 22. Arch Biochem Biophys. 2016. PMID: 26906074 Free PMC article.
-
Mavacamten decreases maximal force and Ca2+ sensitivity in the N47K-myosin regulatory light chain mouse model of hypertrophic cardiomyopathy.Am J Physiol Heart Circ Physiol. 2021 Feb 1;320(2):H881-H890. doi: 10.1152/ajpheart.00345.2020. Epub 2020 Dec 18. Am J Physiol Heart Circ Physiol. 2021. PMID: 33337957 Free PMC article.
-
Functional and Molecular Characterisation of Heart Failure Progression in Mice and the Role of Myosin Regulatory Light Chains in the Recovery of Cardiac Muscle Function.Int J Mol Sci. 2021 Dec 22;23(1):88. doi: 10.3390/ijms23010088. Int J Mol Sci. 2021. PMID: 35008512 Free PMC article.
-
Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains.Pflugers Arch. 2019 May;471(5):683-699. doi: 10.1007/s00424-019-02257-4. Epub 2019 Jan 31. Pflugers Arch. 2019. PMID: 30706179 Free PMC article. Review.
-
N-Terminus of Cardiac Myosin Essential Light Chain Modulates Myosin Step-Size.Biochemistry. 2016 Jan 12;55(1):186-98. doi: 10.1021/acs.biochem.5b00817. Epub 2015 Dec 29. Biochemistry. 2016. PMID: 26671638 Free PMC article.
References
-
- Alpert NR, et al. Molecular mechanics of mouse cardiac myosin isoforms. Am. J. Physiol. Heart Circ. Physiol. 2002;283(4):H1446–H1454. - PubMed
-
- Alpert NR, et al. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. Am. J. Physiol. Heart Circ. Physiol. 2005;288(3):H1097–H1102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
