

Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone
- PMID: 26108478
- PMCID: PMC4479081
- DOI: 10.1186/s40478-015-0214-2
Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone
Abstract
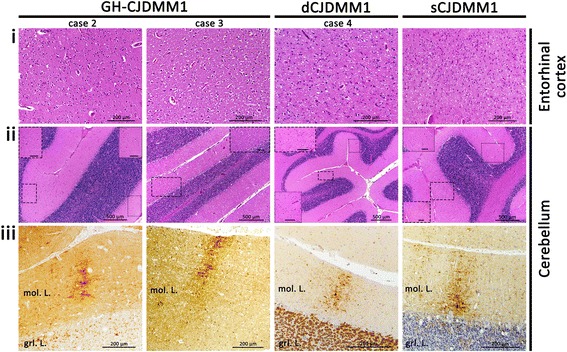
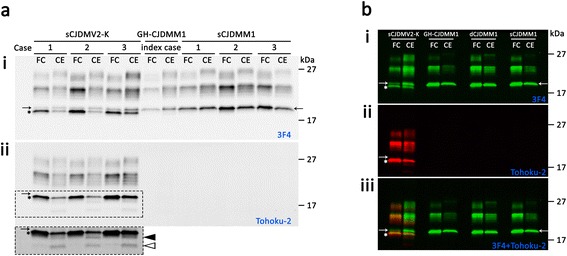
Introduction: The present study compares the clinical, pathological and molecular features of a United States (US) case of growth hormone (GH)-associated Creutzfeldt-Jakob disease (GH-CJD) (index case) to those of two earlier referred US cases of GH-CJD and one case of dura mater (d)-associated CJD (dCJD). All iatrogenic CJD (iCJD) subjects were methionine (M) homozygous at codon 129 (129MM) of the prion protein (PrP) gene and had scrapie prion protein (PrP(Sc)) type 1 (iCJDMM1).
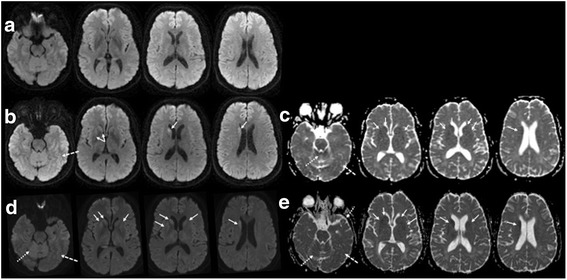
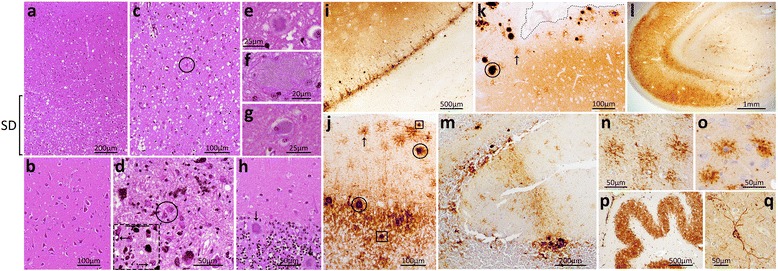
Results: The index subject presented with ataxia, weight loss and changes in the sleep pattern about 38 years after the midpoint of GH treatment. Autopsy examination revealed a neuropathological phenotype reminiscent of both sCJDMV2-K (a sporadic CJD subtype in subjects methionine/valine heterozygous at codon 129 with PrP(Sc) type 2 and the presence of kuru plaques) and variant CJD (vCJD). The two earlier cases of GH-CJDMM1 and the one of dCJDMM1 were associated with neuropathological phenotypes that differed from that of the index case mainly because they lacked PrP plaques. The phenotype of the earlier GH-CJDMM1 cases shared several, but not all, characteristics with sCJDMM1, whereas dCJDMM1 was phenotypically indistinguishable from sCJDMM1. Two distinct groups of dCJDMM1 have also been described in Japan based on clinical features, the presence or absence of PrP plaques and distinct PK-resistant PrP(Sc) (resPrP(Sc)) electrophoretic mobilities. The resPrP(Sc) electrophoretic mobility was, however, identical in our GH-CJDMM1 and dCJDMM1 cases, and matched that of sCJDMM1.
Conclusions: Our study shows that receipt of prion-contaminated GH can lead to a prion disease with molecular features (129MM and PrP(Sc) type 2) and phenotypic characteristics that differ from those of sporadic prion disease (sCJDMM1), a difference that may reflect adaptation of "heterologous" prion strains to the 129MM background.
Figures
Similar articles
-
Classification of sporadic Creutzfeldt-Jakob disease revisited.Brain. 2006 Sep;129(Pt 9):2266-77. doi: 10.1093/brain/awl224. Brain. 2006. PMID: 16923954
-
A novel subtype of sporadic Creutzfeldt-Jakob disease with PRNP codon 129MM genotype and PrP plaques.Acta Neuropathol. 2023 Jul;146(1):121-143. doi: 10.1007/s00401-023-02581-1. Epub 2023 May 8. Acta Neuropathol. 2023. PMID: 37156880 Free PMC article.
-
Neuropathological and biochemical criteria to identify acquired Creutzfeldt-Jakob disease among presumed sporadic cases.Neuropathology. 2016 Jun;36(3):305-10. doi: 10.1111/neup.12270. Epub 2015 Dec 15. Neuropathology. 2016. PMID: 26669818 Review.
-
Atypical Creutzfeldt-Jakob disease with PrP-amyloid plaques in white matter: molecular characterization and transmission to bank voles show the M1 strain signature.Acta Neuropathol Commun. 2017 Nov 23;5(1):87. doi: 10.1186/s40478-017-0496-7. Acta Neuropathol Commun. 2017. PMID: 29169405 Free PMC article.
-
Neuropathology and molecular biology of variant Creutzfeldt-Jakob disease.Curr Top Microbiol Immunol. 2004;284:133-59. doi: 10.1007/978-3-662-08441-0_6. Curr Top Microbiol Immunol. 2004. PMID: 15148991 Review.
Cited by
-
Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: an international study.Acta Neuropathol Commun. 2018 Jan 8;6(1):5. doi: 10.1186/s40478-017-0503-z. Acta Neuropathol Commun. 2018. PMID: 29310723 Free PMC article.
-
Genetics of Prion Disease in Cattle.Bioinform Biol Insights. 2015 Sep 24;9(Suppl 4):1-10. doi: 10.4137/BBI.S29678. eCollection 2015. Bioinform Biol Insights. 2015. PMID: 26462233 Free PMC article. Review.
-
UK Iatrogenic Creutzfeldt-Jakob disease: investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches.Acta Neuropathol. 2017 Apr;133(4):579-595. doi: 10.1007/s00401-016-1638-x. Epub 2016 Nov 3. Acta Neuropathol. 2017. PMID: 27812793 Free PMC article.
-
Cerebellar compartmentation of prion pathogenesis.Brain Pathol. 2018 Mar;28(2):240-263. doi: 10.1111/bpa.12503. Epub 2017 Apr 10. Brain Pathol. 2018. PMID: 28268246 Free PMC article.
-
Human prion diseases: surgical lessons learned from iatrogenic prion transmission.Neurosurg Focus. 2016 Jul;41(1):E10. doi: 10.3171/2016.5.FOCUS15126. Neurosurg Focus. 2016. PMID: 27364252 Free PMC article. Review.
References
-
- Duffy P, Wolf J, Collins G, et al. Letter: possible person-to-person transmission of Creutzfeldt-Jakob disease. N Engl J Med. 1974;290:692–693. - PubMed
-
- Thomas JG, Chenoweth CE, Sullivan SE. Iatrogenic Creutzfeldt-Jakob disease via surgical instruments. J Clin Neurosci Off J Neurosurg Soc Australas. 2013;20:1207–1212. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
