

Association of aberrant DNA methylation in Apc(min/+) mice with the epithelial-mesenchymal transition and Wnt/β-catenin pathways: genome-wide analysis using MeDIP-seq
- PMID: 26101583
- PMCID: PMC4476183
- DOI: 10.1186/s13578-015-0013-2
Association of aberrant DNA methylation in Apc(min/+) mice with the epithelial-mesenchymal transition and Wnt/β-catenin pathways: genome-wide analysis using MeDIP-seq
Abstract
Background: Aberrant DNA methylation at the 5-carbon on cytosine residues (5mC) in CpG dinucleotides is probably the most extensively characterized epigenetic modification in colon cancer. It has been suggested that the loss of adenomatous polyposis coli (APC) function initiates tumorigenesis and that additional genetic and epigenetic events are involved in colon cancer progression. We aimed to study the genome-wide DNA methylation profiles of intestinal tumorigenesis in Apc(min/+) mice.
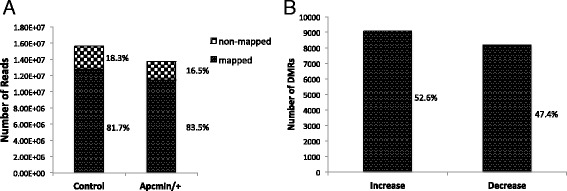
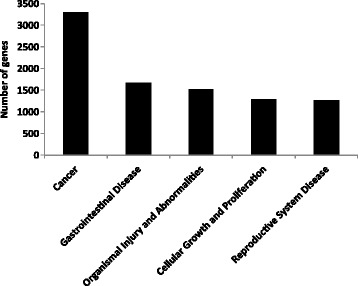
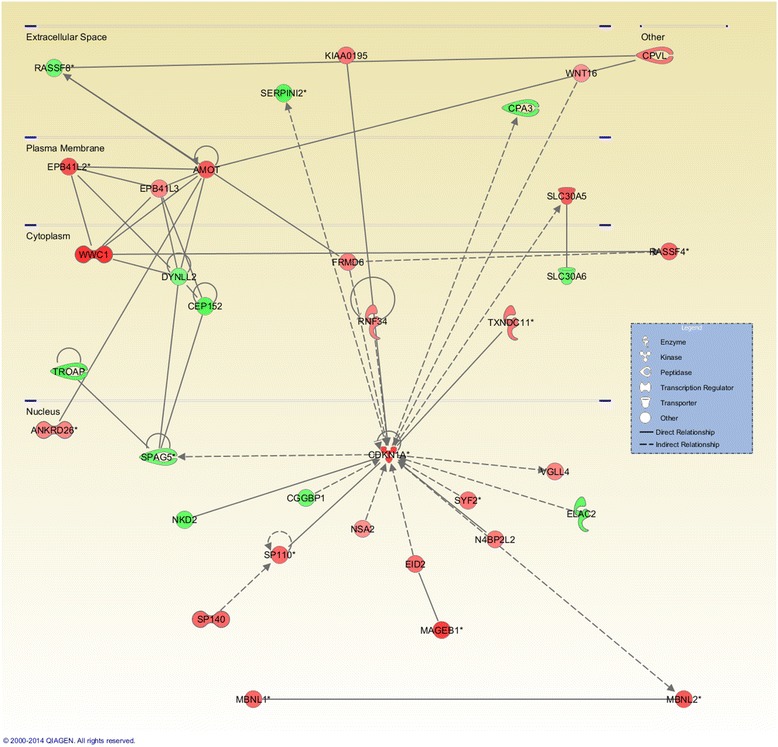
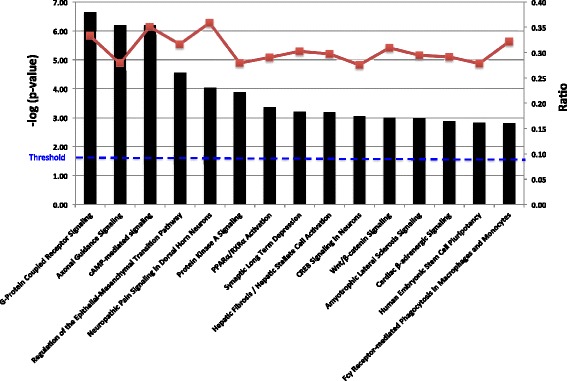
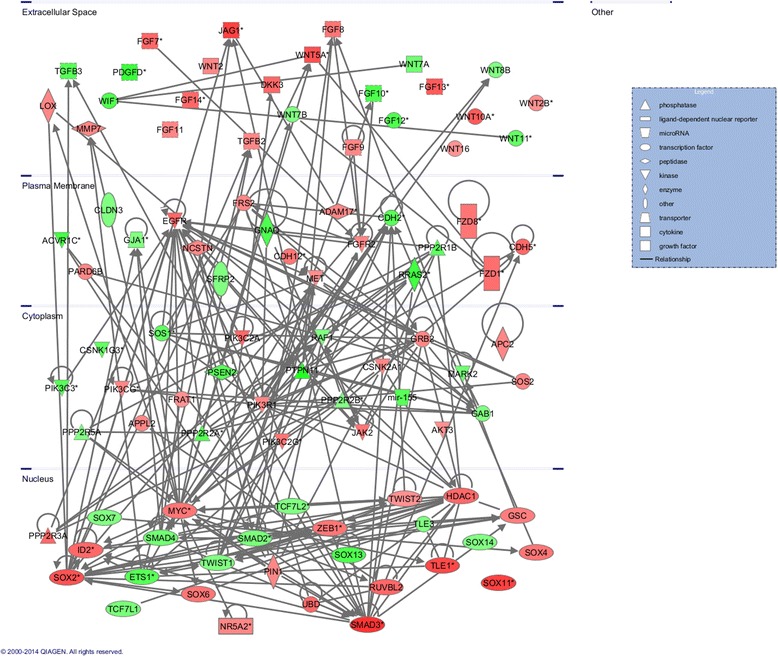
Results: Methylated DNA immunoprecipitation (MeDIP) followed by next-generation sequencing was used to determine the global profile of DNA methylation changes in Apc(min/+) mice. DNA was extracted from adenomatous polyps from Apc(min/+) mice and from normal intestinal tissue from age-matched Apc(+/+) littermates, and the MeDIP-seq assay was performed. Ingenuity Pathway Analysis (IPA) software was used to analyze the data for gene interactions. A total of 17,265 differentially methylated regions (DMRs) displayed a ≥ 2-fold change (log2) in methylation in Apc(min/+) mice; among these DMRs, 9,078 (52.6 %) and 8,187 (47.4 %) exhibited increased and decreased methylation, respectively. Genes with altered methylation patterns were mainly mapped to networks and biological functions associated with cancer and gastrointestinal diseases. Among these networks, several canonical pathways, such as the epithelial-mesenchymal transition (EMT) and Wnt/β-catenin pathways, were significantly associated with genome-wide methylation changes in polyps from Apc(min/+) mice. The identification of certain differentially methylated molecules in the EMT and Wnt/β-catenin pathways, such as APC2 (adenomatosis polyposis coli 2), SFRP2 (secreted frizzled-related protein 2), and DKK3 (dickkopf-related protein 3), was consistent with previous publications.
Conclusions: Our findings indicated that Apc(min/+) mice exhibited extensive aberrant DNA methylation that affected certain signaling pathways, such as the EMT and Wnt/β-catenin pathways. The genome-wide DNA methylation profile of Apc(min/+) mice is informative for future studies investigating epigenetic gene regulation in colon tumorigenesis and the prevention of colon cancer.
Keywords: DNA methylation; Epigenetic; Epithelial-mesenchymal transition pathway; MeDIP-seq; Wnt/β-catenin pathway.
Figures
Similar articles
-
Epigenetic alterations of CDH1 and APC genes: relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast.Life Sci. 2008 Aug 29;83(9-10):318-25. doi: 10.1016/j.lfs.2008.06.019. Epub 2008 Jul 1. Life Sci. 2008. PMID: 18662704
-
Epigenetic alterations in TRAMP mice: epigenome DNA methylation profiling using MeDIP-seq.Cell Biosci. 2018 Jan 12;8:3. doi: 10.1186/s13578-018-0201-y. eCollection 2018. Cell Biosci. 2018. PMID: 29344347 Free PMC article.
-
Elevated Dnmt3a activity promotes polyposis in Apc(Min) mice by relaxing extracellular restraints on Wnt signaling.Gastroenterology. 2009 Sep;137(3):902-13, 913.e1-11. doi: 10.1053/j.gastro.2009.05.042. Epub 2009 May 18. Gastroenterology. 2009. PMID: 19454286
-
Gene methylation in gastric cancer.Clin Chim Acta. 2013 Sep 23;424:53-65. doi: 10.1016/j.cca.2013.05.002. Epub 2013 May 10. Clin Chim Acta. 2013. PMID: 23669186 Review.
-
Interaction of the Wnt/β-catenin and RAS-ERK pathways involving co-stabilization of both β-catenin and RAS plays important roles in the colorectal tumorigenesis.Adv Biol Regul. 2018 May;68:46-54. doi: 10.1016/j.jbior.2018.01.001. Epub 2018 Jan 10. Adv Biol Regul. 2018. PMID: 29449169 Review.
Cited by
-
Epigenetics/epigenomics and prevention by curcumin of early stages of inflammatory-driven colon cancer.Mol Carcinog. 2020 Feb;59(2):227-236. doi: 10.1002/mc.23146. Epub 2019 Dec 9. Mol Carcinog. 2020. PMID: 31820492 Free PMC article. Review.
-
A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC.Nat Commun. 2016 Jun 15;7:11743. doi: 10.1038/ncomms11743. Nat Commun. 2016. PMID: 27302369 Free PMC article.
-
Mouse Models for Application in Colorectal Cancer: Understanding the Pathogenesis and Relevance to the Human Condition.Biomedicines. 2022 Jul 15;10(7):1710. doi: 10.3390/biomedicines10071710. Biomedicines. 2022. PMID: 35885015 Free PMC article. Review.
-
Loss of FFAR2 promotes colon cancer by epigenetic dysregulation of inflammation suppressors.Int J Cancer. 2018 Aug 15;143(4):886-896. doi: 10.1002/ijc.31366. Epub 2018 Mar 30. Int J Cancer. 2018. PMID: 29524208 Free PMC article.
-
In silico identification of novel biomarkers for key players in transition from normal colon tissue to adenomatous polyps.PLoS One. 2022 Apr 29;17(4):e0267973. doi: 10.1371/journal.pone.0267973. eCollection 2022. PLoS One. 2022. PMID: 35486660 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
