

MOST+: A de novo motif finding approach combining genomic sequence and heterogeneous genome-wide signatures
- PMID: 26099518
- PMCID: PMC4474412
- DOI: 10.1186/1471-2164-16-S7-S13
MOST+: A de novo motif finding approach combining genomic sequence and heterogeneous genome-wide signatures
Abstract
Background: Motifs are regulatory elements that will activate or inhibit the expression of related genes when proteins (such as transcription factors, TFs) bind to them. Therefore, motif finding is important to understand the mechanisms of gene regulation. De novo discovery of regulatory elements, like transcription factor binding sites (TFBSs), has long been a major challenge to gain insight on mechanisms of gene regulation. Recent advances in experimental profiling of genome-wide signals such as histone modifications and DNase I hypersensitivity sites allow scientists to develop better computational methods to enhance motif discovery. However, existing methods for motif finding suffer from high false positive rates and slow speed, and it's difficult to evaluate the performance of these methods systematically.
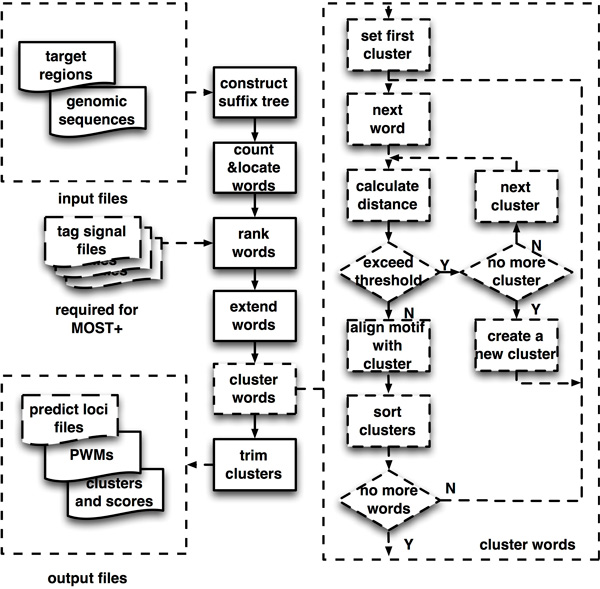
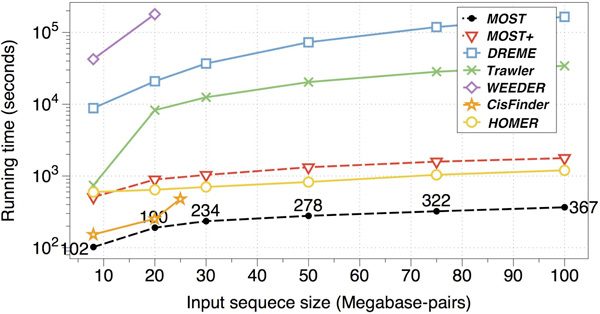
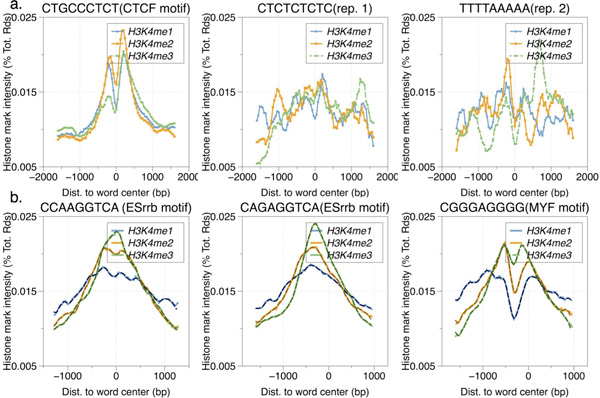
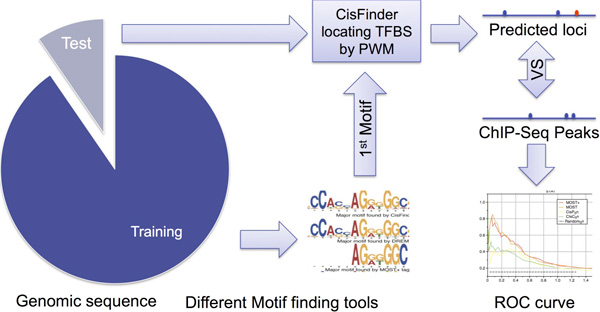
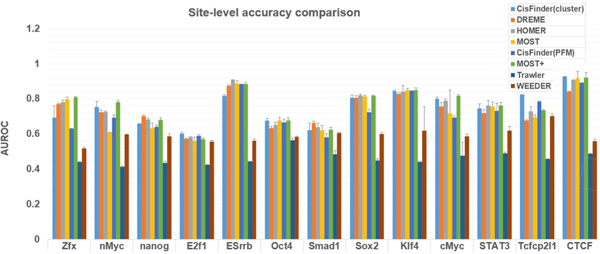
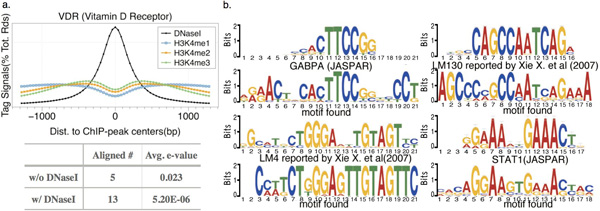
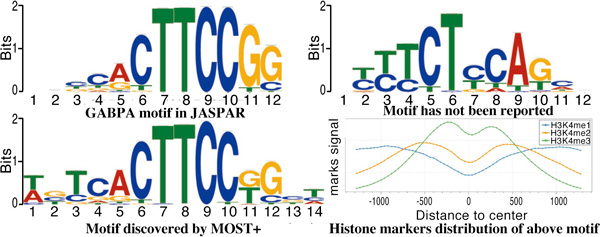
Result: Here we present MOST+, a motif finder integrating genomic sequences and genome-wide signals such as intensity and shape features from histone modification marks and DNase I hypersensitivity sites, to improve the prediction accuracy. MOST+ can detect motifs from a large input sequence of about 100 Mbs within a few minutes. Systematic comparison method has been established and MOST+ has been compared with existing methods.
Conclusion: MOST+ is a fast and accurate de novo method for motif finding by integrating genomic sequence and experimental signals as clues.
Figures
Similar articles
-
Genome-wide prediction of transcriptional regulatory elements of human promoters using gene expression and promoter analysis data.BMC Bioinformatics. 2006 Jul 4;7:330. doi: 10.1186/1471-2105-7-330. BMC Bioinformatics. 2006. PMID: 16817975 Free PMC article.
-
GSMC: Combining Parallel Gibbs Sampling with Maximal Cliques for Hunting DNA Motif.J Comput Biol. 2017 Dec;24(12):1243-1253. doi: 10.1089/cmb.2017.0100. Epub 2017 Nov 8. J Comput Biol. 2017. PMID: 29116820 Free PMC article.
-
Identification of context-dependent motifs by contrasting ChIP binding data.Bioinformatics. 2010 Nov 15;26(22):2826-32. doi: 10.1093/bioinformatics/btq546. Epub 2010 Sep 23. Bioinformatics. 2010. PMID: 20870645 Free PMC article.
-
Computational identification of transcriptional regulatory elements in DNA sequence.Nucleic Acids Res. 2006 Jul 19;34(12):3585-98. doi: 10.1093/nar/gkl372. Print 2006. Nucleic Acids Res. 2006. PMID: 16855295 Free PMC article. Review.
-
A comparative study on computational two-block motif detection: algorithms and applications.Mol Pharm. 2008 Jan-Feb;5(1):3-16. doi: 10.1021/mp7001126. Epub 2007 Dec 13. Mol Pharm. 2008. PMID: 18076137 Review.
Cited by
-
Computational modeling of in vivo and in vitro protein-DNA interactions by multiple instance learning.Bioinformatics. 2017 Jul 15;33(14):2097-2105. doi: 10.1093/bioinformatics/btx115. Bioinformatics. 2017. PMID: 28334224 Free PMC article.
-
Education, collaboration, and innovation: intelligent biology and medicine in the era of big data.BMC Genomics. 2015;16 Suppl 7(Suppl 7):S1. doi: 10.1186/1471-2164-16-S7-S1. Epub 2015 Jun 11. BMC Genomics. 2015. PMID: 26099197 Free PMC article.
References
-
- Bailey TL, Elkan C. Unsupervised Learning of Multiple Motifs in Biopolymers Using Expectation Maximization. University of California San Diego. Dept.of Computer Science and Engineering; 1993.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
