

Engineered CRISPR-Cas9 nucleases with altered PAM specificities
- PMID: 26098369
- PMCID: PMC4540238
- DOI: 10.1038/nature14592
Engineered CRISPR-Cas9 nucleases with altered PAM specificities
Abstract
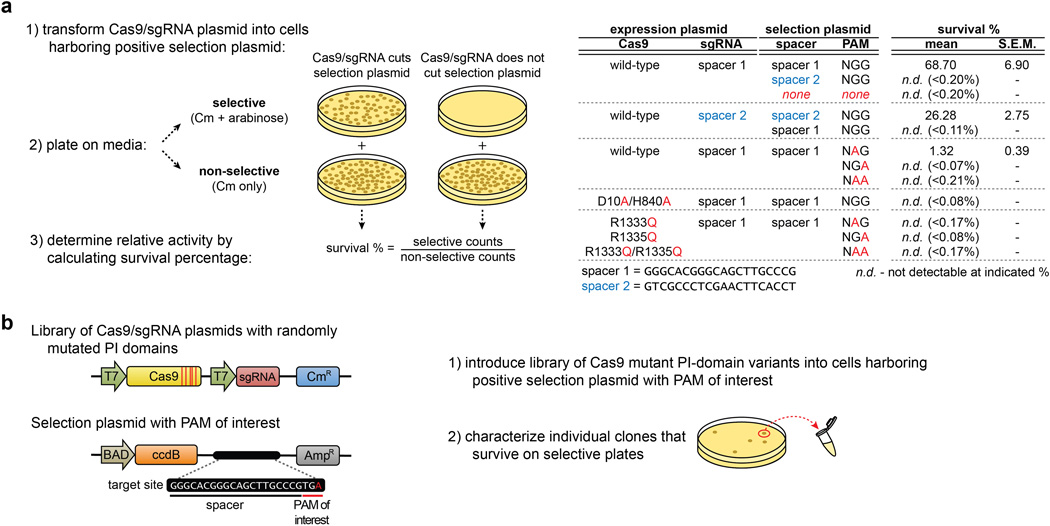
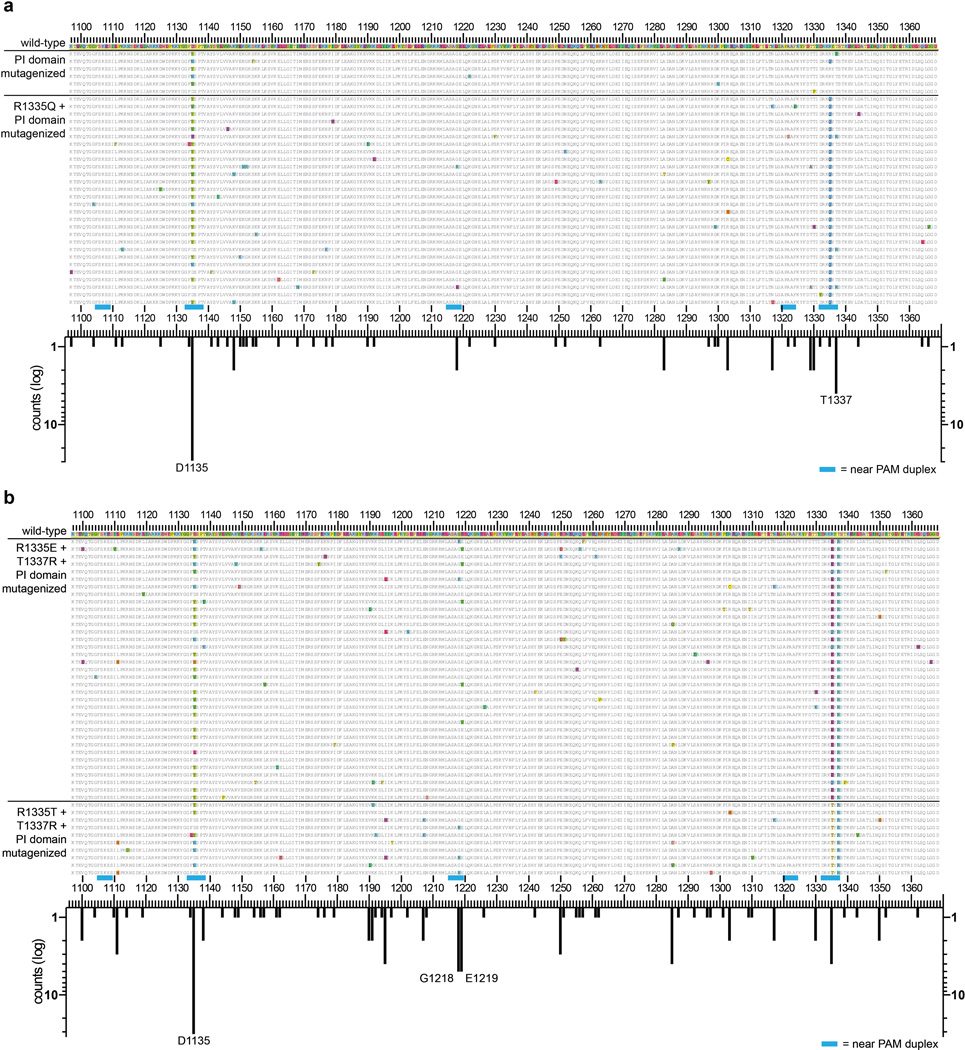
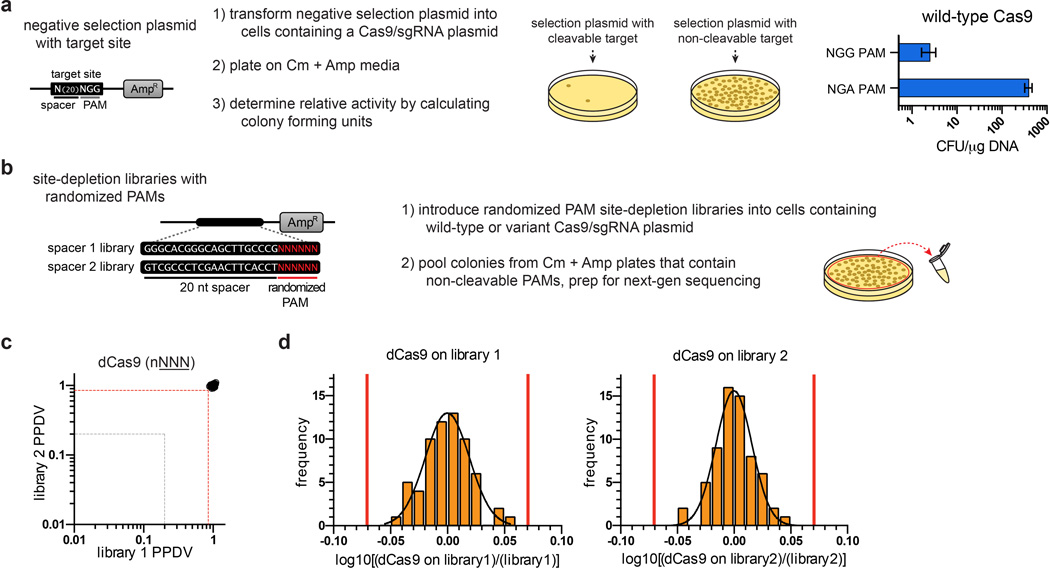
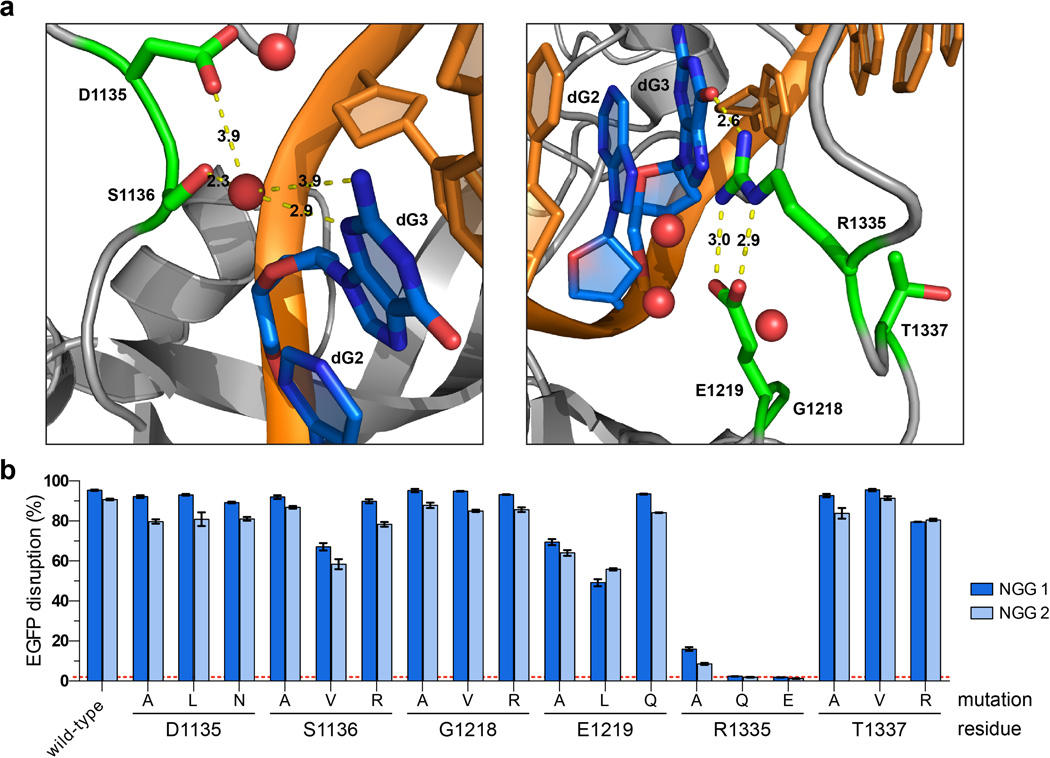
Although CRISPR-Cas9 nucleases are widely used for genome editing, the range of sequences that Cas9 can recognize is constrained by the need for a specific protospacer adjacent motif (PAM). As a result, it can often be difficult to target double-stranded breaks (DSBs) with the precision that is necessary for various genome-editing applications. The ability to engineer Cas9 derivatives with purposefully altered PAM specificities would address this limitation. Here we show that the commonly used Streptococcus pyogenes Cas9 (SpCas9) can be modified to recognize alternative PAM sequences using structural information, bacterial selection-based directed evolution, and combinatorial design. These altered PAM specificity variants enable robust editing of endogenous gene sites in zebrafish and human cells not currently targetable by wild-type SpCas9, and their genome-wide specificities are comparable to wild-type SpCas9 as judged by GUIDE-seq analysis. In addition, we identify and characterize another SpCas9 variant that exhibits improved specificity in human cells, possessing better discrimination against off-target sites with non-canonical NAG and NGA PAMs and/or mismatched spacers. We also find that two smaller-size Cas9 orthologues, Streptococcus thermophilus Cas9 (St1Cas9) and Staphylococcus aureus Cas9 (SaCas9), function efficiently in the bacterial selection systems and in human cells, suggesting that our engineering strategies could be extended to Cas9s from other species. Our findings provide broadly useful SpCas9 variants and, more importantly, establish the feasibility of engineering a wide range of Cas9s with altered and improved PAM specificities.
Conflict of interest statement
Conflict of interest statement: J.K.J. is a consultant for Horizon Discovery. J.K.J. has financial interests in Editas Medicine, Hera Testing Laboratories, Poseida Therapeutics, and Transposagen Biopharmaceuticals. J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Figures





Comment in
-
Engineering Cas9.Nat Methods. 2015 Aug;12(8):709. doi: 10.1038/nmeth.3514. Nat Methods. 2015. PMID: 26451425 No abstract available.
Similar articles
-
High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects.Nature. 2016 Jan 28;529(7587):490-5. doi: 10.1038/nature16526. Epub 2016 Jan 6. Nature. 2016. PMID: 26735016 Free PMC article.
-
Cas9 specifies functional viral targets during CRISPR-Cas adaptation.Nature. 2015 Mar 12;519(7542):199-202. doi: 10.1038/nature14245. Epub 2015 Feb 18. Nature. 2015. PMID: 25707807 Free PMC article.
-
Evolved Cas9 variants with broad PAM compatibility and high DNA specificity.Nature. 2018 Apr 5;556(7699):57-63. doi: 10.1038/nature26155. Epub 2018 Feb 28. Nature. 2018. PMID: 29512652 Free PMC article.
-
Cas9, Cpf1 and C2c1/2/3-What's next?Bioengineered. 2017 May 4;8(3):265-273. doi: 10.1080/21655979.2017.1282018. Epub 2017 Jan 31. Bioengineered. 2017. PMID: 28140746 Free PMC article. Review.
-
Application of CRISPR/Cas9 genome editing to the study and treatment of disease.Arch Toxicol. 2015 Jul;89(7):1023-34. doi: 10.1007/s00204-015-1504-y. Epub 2015 Apr 1. Arch Toxicol. 2015. PMID: 25827103 Review.
Cited by
-
How to Completely Squeeze a Fungus-Advanced Genome Mining Tools for Novel Bioactive Substances.Pharmaceutics. 2022 Aug 31;14(9):1837. doi: 10.3390/pharmaceutics14091837. Pharmaceutics. 2022. PMID: 36145585 Free PMC article. Review.
-
Retinal gene therapy: an eye-opener of the 21st century.Gene Ther. 2021 May;28(5):209-216. doi: 10.1038/s41434-020-0168-2. Epub 2020 Jun 19. Gene Ther. 2021. PMID: 32561864 No abstract available.
-
A more efficient CRISPR-Cas12a variant derived from Lachnospiraceae bacterium MA2020.Mol Ther Nucleic Acids. 2021 Feb 18;24:40-53. doi: 10.1016/j.omtn.2021.02.012. eCollection 2021 Jun 4. Mol Ther Nucleic Acids. 2021. PMID: 33738137 Free PMC article.
-
Efficient genome engineering approaches for the short-lived African turquoise killifish.Nat Protoc. 2016 Oct;11(10):2010-2028. doi: 10.1038/nprot.2016.103. Epub 2016 Sep 22. Nat Protoc. 2016. PMID: 27658015
-
Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors.Nat Biotechnol. 2020 Jul;38(7):824-844. doi: 10.1038/s41587-020-0561-9. Epub 2020 Jun 22. Nat Biotechnol. 2020. PMID: 32572269 Review.
References
-
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. - PubMed
-
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
