

Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of "de novo" SCN1A Mutations in Children with Dravet Syndrome
- PMID: 26096185
- PMCID: PMC5034833
- DOI: 10.1002/humu.22819
Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of "de novo" SCN1A Mutations in Children with Dravet Syndrome
Abstract
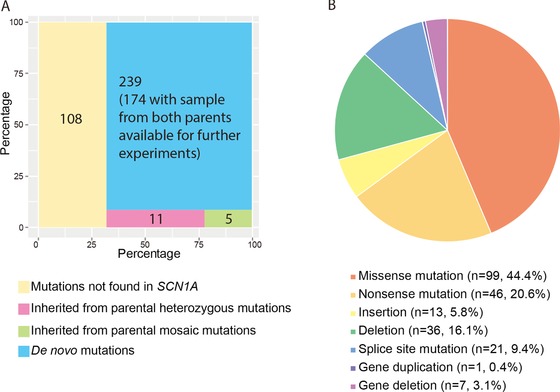
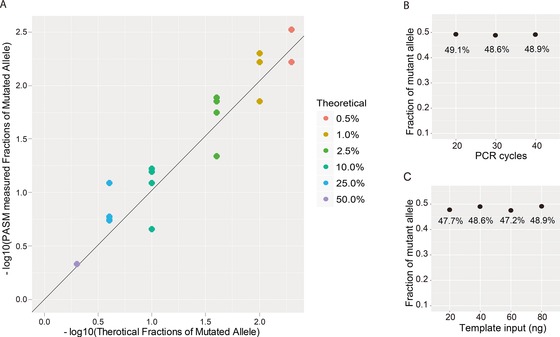
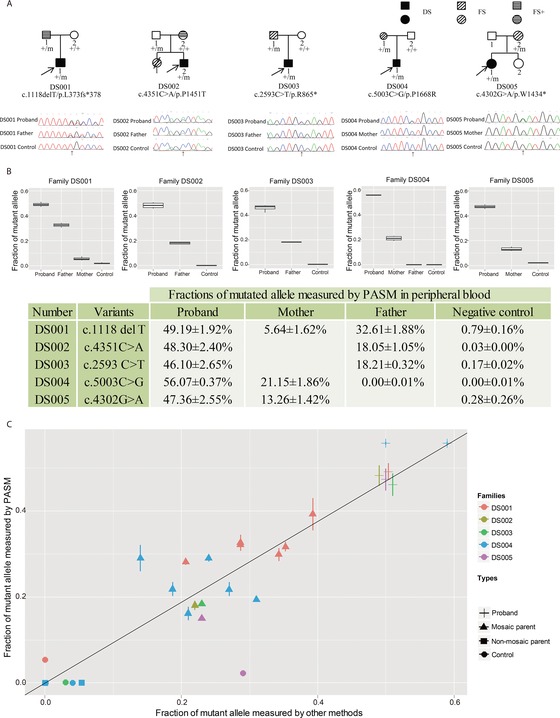
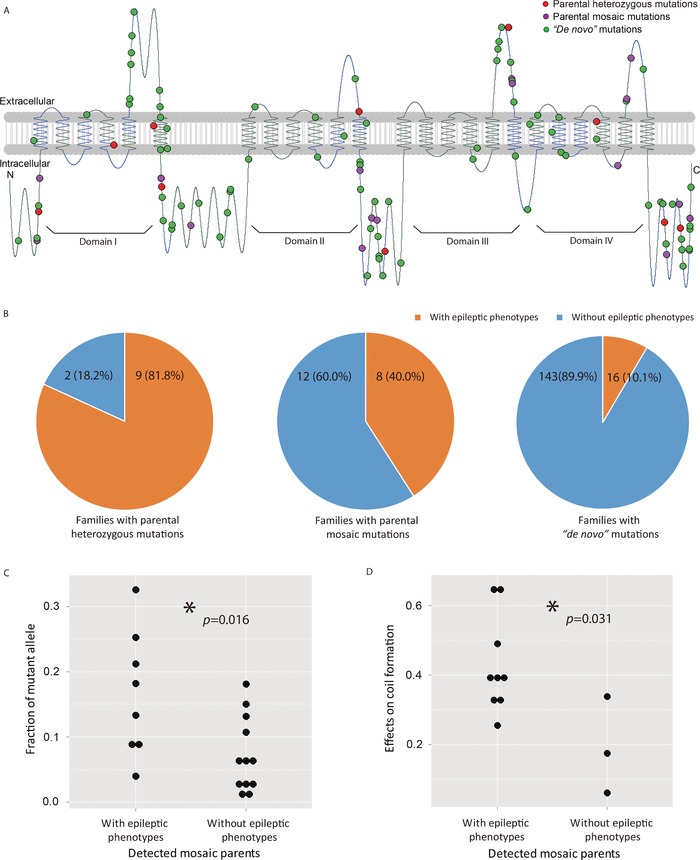
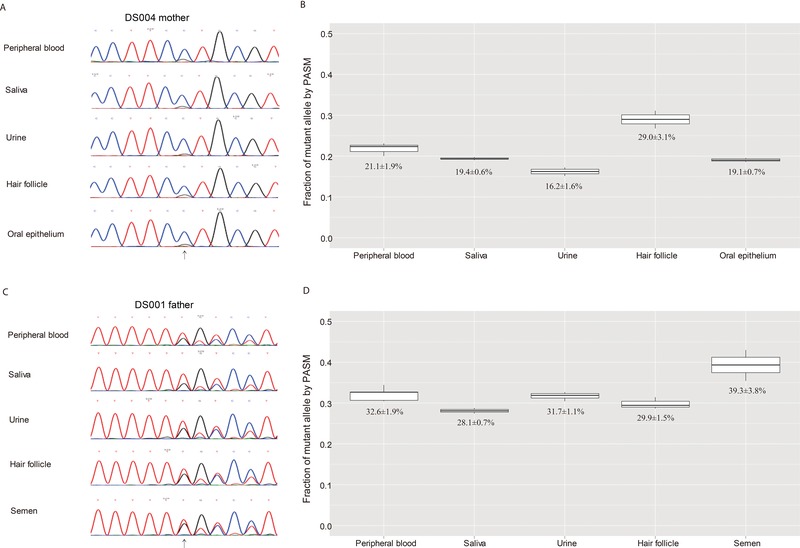
The majority of children with Dravet syndrome (DS) are caused by de novo SCN1A mutations. To investigate the origin of the mutations, we developed and applied a new method that combined deep amplicon resequencing with a Bayesian model to detect and quantify allelic fractions with improved sensitivity. Of 174 SCN1A mutations in DS probands which were considered "de novo" by Sanger sequencing, we identified 15 cases (8.6%) of parental mosaicism. We identified another five cases of parental mosaicism that were also detectable by Sanger sequencing. Fraction of mutant alleles in the 20 cases of parental mosaicism ranged from 1.1% to 32.6%. Thirteen (65% of 20) mutations originated paternally and seven (35% of 20) maternally. Twelve (60% of 20) mosaic parents did not have any epileptic symptoms. Their mutant allelic fractions were significantly lower than those in mosaic parents with epileptic symptoms (P = 0.016). We identified mosaicism with varied allelic fractions in blood, saliva, urine, hair follicle, oral epithelium, and semen, demonstrating that postzygotic mutations could affect multiple somatic cells as well as germ cells. Our results suggest that more sensitive tools for detecting low-level mosaicism in parents of families with seemingly "de novo" mutations will allow for better informed genetic counseling.
Keywords: Dravet syndrome; SCN1A; de novo; mosaic; next-generation sequencing; somatic mutation.
© 2015 The Authors. **Human Mutation published by Wiley Periodicals, Inc.
Figures
Similar articles
-
[Study on mosaicism of SCN1A gene mutation in parents of children with Dravet syndrome].Zhonghua Er Ke Za Zhi. 2017 Nov 2;55(11):818-823. doi: 10.3760/cma.j.issn.0578-1310.2017.11.005. Zhonghua Er Ke Za Zhi. 2017. PMID: 29141311 Chinese.
-
[Analysis of parental origin of de novo SCN1A mutations in Dravet syndrome].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015 Aug;32(4):457-61. doi: 10.3760/cma.j.issn.1003-9406.2015.04.001. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015. PMID: 26252084 Chinese.
-
Parental mosaicism in another case of Dravet syndrome caused by a novel SCN1A deletion: a case report.J Med Case Rep. 2016 Mar 29;10:67. doi: 10.1186/s13256-016-0854-2. J Med Case Rep. 2016. PMID: 27021235 Free PMC article. Review.
-
Somatic mosaic deletions involving SCN1A cause Dravet syndrome.Am J Med Genet A. 2018 Mar;176(3):657-662. doi: 10.1002/ajmg.a.38596. Epub 2018 Jan 17. Am J Med Genet A. 2018. PMID: 29341473
-
Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies.Epilepsia. 2019 Dec;60 Suppl 3:S2-S7. doi: 10.1111/epi.16054. Epilepsia. 2019. PMID: 31904125 Review.
Cited by
-
Mosaic PKHD1 in Polycystic Kidneys Caused Aberrant Protein Expression in the Mitochondria and Lysosomes.Front Med (Lausanne). 2021 Dec 16;8:743150. doi: 10.3389/fmed.2021.743150. eCollection 2021. Front Med (Lausanne). 2021. PMID: 34977057 Free PMC article.
-
Clinical Application of Epilepsy Genetics in Africa: Is Now the Time?Front Neurol. 2018 May 2;9:276. doi: 10.3389/fneur.2018.00276. eCollection 2018. Front Neurol. 2018. PMID: 29770117 Free PMC article.
-
eHealth as a Facilitator of Precision Medicine in Epilepsy.Biomed Hub. 2017 Nov 21;2(Suppl 1):137-145. doi: 10.1159/000481793. eCollection 2017 Nov-Dec. Biomed Hub. 2017. PMID: 31988944 Free PMC article. Review.
-
High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders.Genet Med. 2018 Apr;20(4):403-410. doi: 10.1038/gim.2017.114. Epub 2017 Aug 24. Genet Med. 2018. PMID: 28837158 Free PMC article.
-
A Case Report of Parental Germline Mosaicism in the PCDH19 Gene of Two Iranian Siblings.Basic Clin Neurosci. 2024 Jul-Aug;15(4):541-552. doi: 10.32598/bcn.2023.5507.1. Epub 2024 Jul 1. Basic Clin Neurosci. 2024. PMID: 39553263 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
