

Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs
- PMID: 26024389
- PMCID: PMC4816114
- DOI: 10.1038/cdd.2015.59
Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs
Abstract
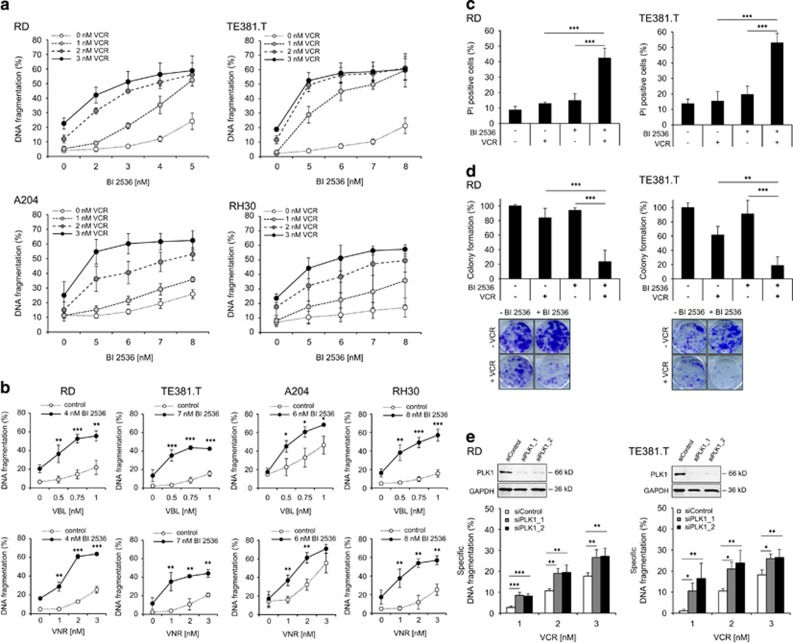
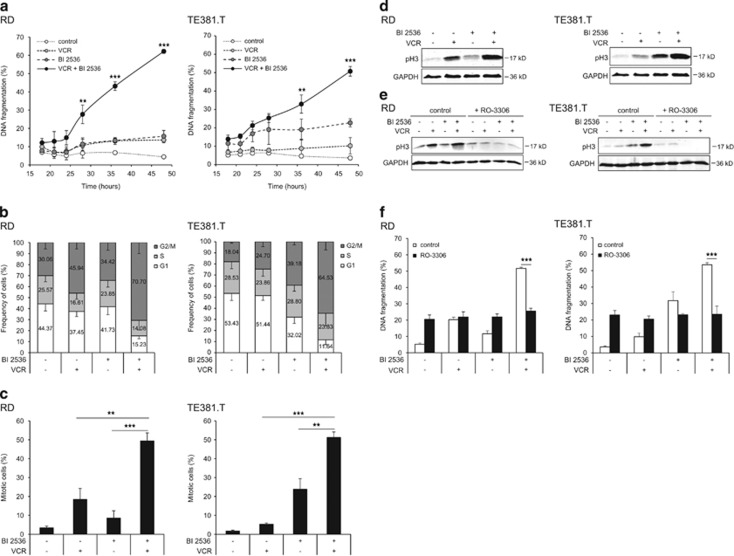
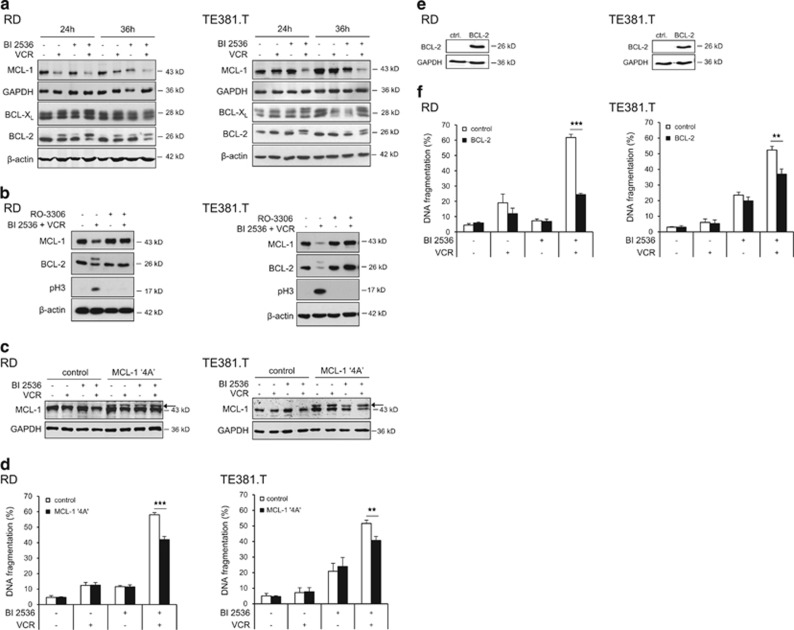
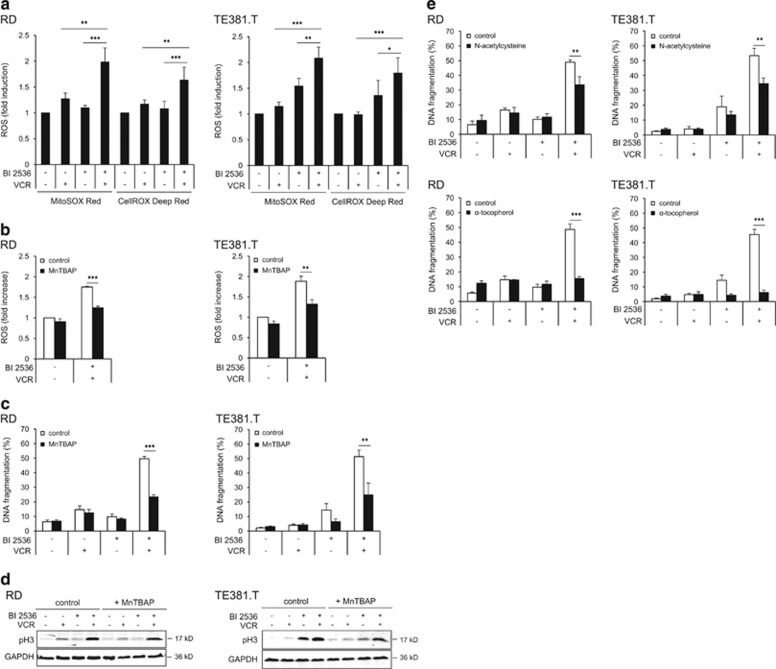
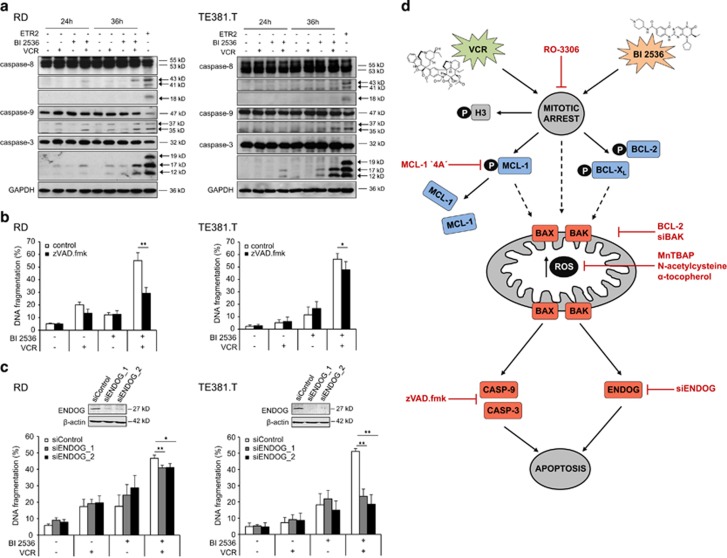
Polo-like kinase 1 (PLK1) is frequently overexpressed in cancer, which correlates with poor prognosis. Therefore, we investigated PLK1 as therapeutic target using rhabdomyosarcoma (RMS) as a model. Here, we identify a novel synthetic lethal interaction of PLK1 inhibitors and microtubule-destabilizing drugs in preclinical RMS models and elucidate the underlying molecular mechanisms of this synergism. PLK1 inhibitors (i.e., BI 2536 and BI 6727) synergistically induce apoptosis together with microtubule-destabilizing drugs (i.e., vincristine (VCR), vinblastine (VBL) and vinorelbine (VNR)) in several RMS cell lines (combination index <0.9) including a patient-derived primary RMS culture. Importantly, PLK1 inhibitors and VCR cooperate to significantly suppress RMS growth in two in vivo models, including a mouse xenograft model, without causing additive toxicity. In addition, no toxicity was observed in non-malignant fibroblast or myoblast cultures. Mechanistically, BI 2536/VCR co-treatment triggers mitotic arrest, which initiates mitochondrial apoptosis by inactivation of antiapoptotic BCL-2 family proteins, followed by BAX/BAK activation, production of reactive oxygen species (ROS) and activation of caspase-dependent or caspase-independent effector pathways. This conclusion is supported by data showing that BI 2536/VCR-induced apoptosis is significantly inhibited by preventing cells to enter mitosis, by overexpression of BCL-2 or a non-degradable MCL-1 mutant, by BAK knockdown, ROS scavengers, caspase inhibition or endonuclease G silencing. This identification of a novel synthetic lethality of PLK1 inhibitors and microtubule-destabilizing drugs has important implications for developing PLK1 inhibitor-based combination treatments.
Figures


Similar articles
-
Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma.Cancer Lett. 2015 Aug 28;365(1):37-46. doi: 10.1016/j.canlet.2015.04.011. Epub 2015 Apr 23. Cancer Lett. 2015. PMID: 25917079
-
Synergistic induction of apoptosis by a polo-like kinase 1 inhibitor and microtubule-interfering drugs in Ewing sarcoma cells.Int J Cancer. 2016 Jan 15;138(2):497-506. doi: 10.1002/ijc.29725. Epub 2015 Sep 1. Int J Cancer. 2016. PMID: 26260582
-
Polo-like kinase 1 inhibition sensitizes neuroblastoma cells for vinca alkaloid-induced apoptosis.Oncotarget. 2016 Feb 23;7(8):8700-11. doi: 10.18632/oncotarget.3901. Oncotarget. 2016. PMID: 26046302 Free PMC article.
-
BI_2536--targeting the mitotic kinase Polo-like kinase 1 (Plk1).Recent Results Cancer Res. 2010;184:215-8. doi: 10.1007/978-3-642-01222-8_15. Recent Results Cancer Res. 2010. PMID: 20072841 Review.
-
Inhibitors of the Polo-Box Domain of Polo-Like Kinase 1.Chembiochem. 2016 Apr 15;17(8):650-6. doi: 10.1002/cbic.201500580. Epub 2016 Jan 29. Chembiochem. 2016. PMID: 26662918 Review.
Cited by
-
A Perspective on Polo-Like Kinase-1 Inhibition for the Treatment of Rhabdomyosarcomas.Front Oncol. 2019 Nov 22;9:1271. doi: 10.3389/fonc.2019.01271. eCollection 2019. Front Oncol. 2019. PMID: 31824851 Free PMC article. Review.
-
CYT997(Lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced ER stress and ROS in osteosarcoma.J Exp Clin Cancer Res. 2019 Jan 31;38(1):44. doi: 10.1186/s13046-019-1047-9. J Exp Clin Cancer Res. 2019. PMID: 30704503 Free PMC article.
-
BRCA1 orchestrates the response to BI-2536 and its combination with alisertib in MYC-driven small cell lung cancer.Cell Death Dis. 2024 Jul 31;15(7):551. doi: 10.1038/s41419-024-06950-w. Cell Death Dis. 2024. PMID: 39085197 Free PMC article.
-
Cell cycle arrest in mitosis promotes interferon-induced necroptosis.Cell Death Differ. 2019 Oct;26(10):2046-2060. doi: 10.1038/s41418-019-0298-5. Epub 2019 Feb 11. Cell Death Differ. 2019. PMID: 30742091 Free PMC article.
-
Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery.Cancer. 2019 Mar 1;125(5):712-725. doi: 10.1002/cncr.31837. Epub 2018 Nov 27. Cancer. 2019. PMID: 30480765 Free PMC article.
References
-
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006; 25: 4798–4811. - PubMed
-
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9: 447–464. - PubMed
-
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432: 307–315. - PubMed
-
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26: 7773–7779. - PubMed
-
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9: 643–660. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
