

A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data
- PMID: 26015273
- PMCID: PMC4444969
- DOI: 10.1038/srep10576
A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data
Abstract
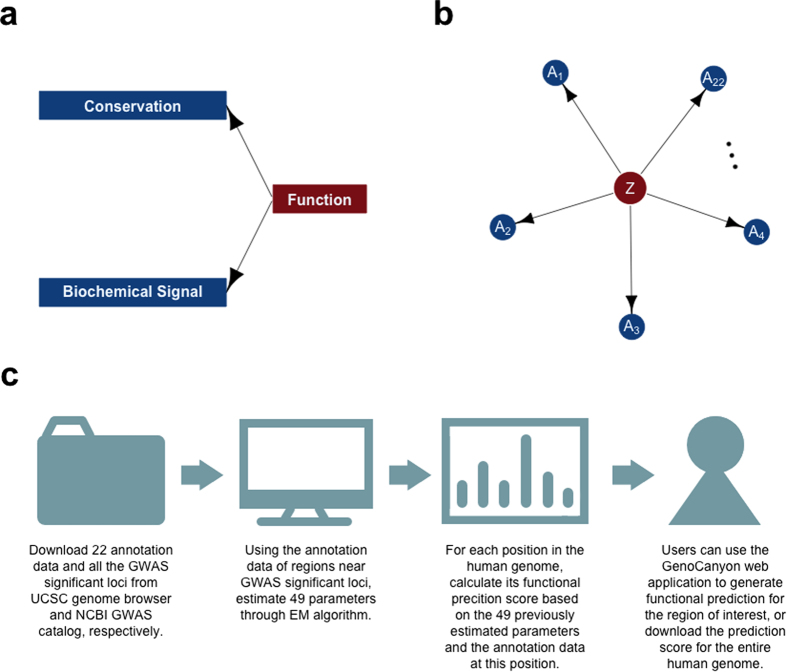
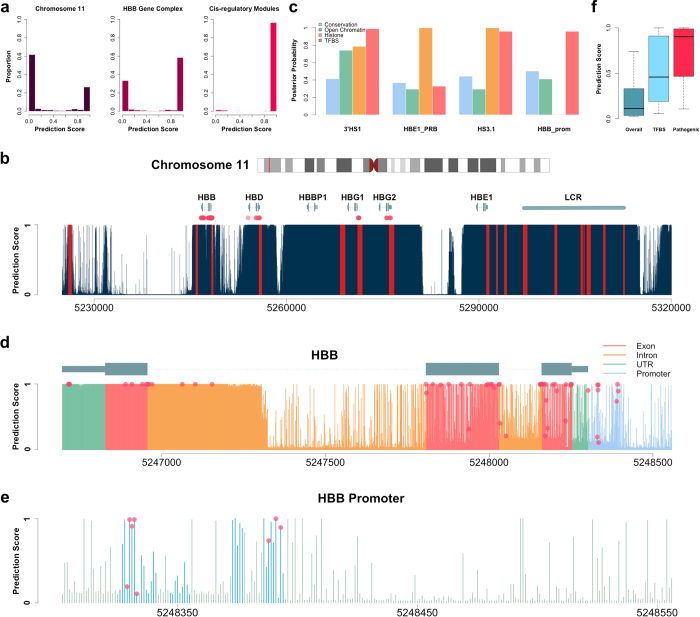
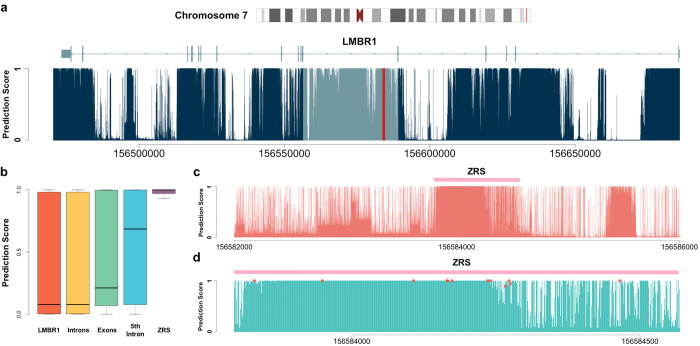
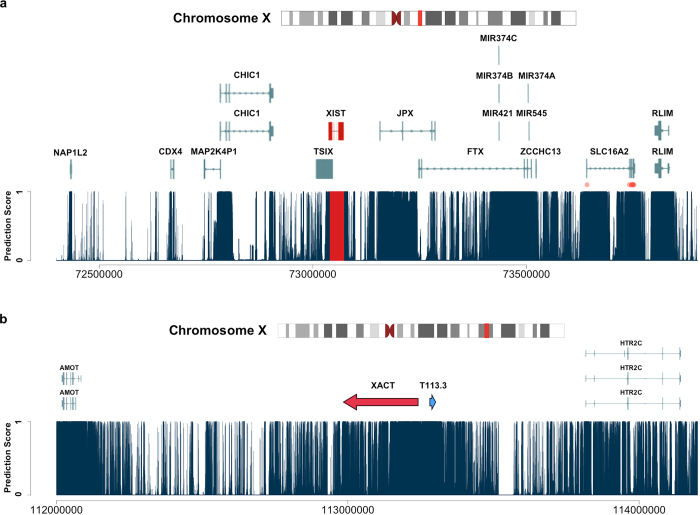
Identifying functional regions in the human genome is a major goal in human genetics. Great efforts have been made to functionally annotate the human genome either through computational predictions, such as genomic conservation, or high-throughput experiments, such as the ENCODE project. These efforts have resulted in a rich collection of functional annotation data of diverse types that need to be jointly analyzed for integrated interpretation and annotation. Here we present GenoCanyon, a whole-genome annotation method that performs unsupervised statistical learning using 22 computational and experimental annotations thereby inferring the functional potential of each position in the human genome. With GenoCanyon, we are able to predict many of the known functional regions. The ability of predicting functional regions as well as its generalizable statistical framework makes GenoCanyon a unique and powerful tool for whole-genome annotation. The GenoCanyon web server is available at http://genocanyon.med.yale.edu.
Figures
Similar articles
-
Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer's disease.PLoS Genet. 2017 Jul 24;13(7):e1006933. doi: 10.1371/journal.pgen.1006933. eCollection 2017 Jul. PLoS Genet. 2017. PMID: 28742084 Free PMC article.
-
Integrative Tissue-Specific Functional Annotations in the Human Genome Provide Novel Insights on Many Complex Traits and Improve Signal Prioritization in Genome Wide Association Studies.PLoS Genet. 2016 Apr 8;12(4):e1005947. doi: 10.1371/journal.pgen.1005947. eCollection 2016 Apr. PLoS Genet. 2016. PMID: 27058395 Free PMC article.
-
[Analysis, identification and correction of some errors of model refseqs appeared in NCBI Human Gene Database by in silico cloning and experimental verification of novel human genes].Yi Chuan Xue Bao. 2004 May;31(5):431-43. Yi Chuan Xue Bao. 2004. PMID: 15478601 Chinese.
-
Current trend of annotating single nucleotide variation in humans--A case study on SNVrap.Methods. 2015 Jun;79-80:32-40. doi: 10.1016/j.ymeth.2014.10.003. Epub 2014 Oct 13. Methods. 2015. PMID: 25308971 Review.
-
dbWGFP: a database and web server of human whole-genome single nucleotide variants and their functional predictions.Database (Oxford). 2016 Mar 17;2016:baw024. doi: 10.1093/database/baw024. Print 2016. Database (Oxford). 2016. PMID: 26989155 Free PMC article.
Cited by
-
Region-based analysis with functional annotation identifies genes associated with cognitive function in South Asians from India.medRxiv [Preprint]. 2024 Jan 18:2024.01.18.24301482. doi: 10.1101/2024.01.18.24301482. medRxiv. 2024. PMID: 38293024 Free PMC article. Preprint.
-
Computational Methods for the Pharmacogenetic Interpretation of Next Generation Sequencing Data.Front Pharmacol. 2018 Dec 4;9:1437. doi: 10.3389/fphar.2018.01437. eCollection 2018. Front Pharmacol. 2018. PMID: 30564131 Free PMC article. Review.
-
Improving SNP prioritization and pleiotropic architecture estimation by incorporating prior knowledge using graph-GPA.Bioinformatics. 2018 Jun 15;34(12):2139-2141. doi: 10.1093/bioinformatics/bty061. Bioinformatics. 2018. PMID: 29432514 Free PMC article.
-
VarCards: an integrated genetic and clinical database for coding variants in the human genome.Nucleic Acids Res. 2018 Jan 4;46(D1):D1039-D1048. doi: 10.1093/nar/gkx1039. Nucleic Acids Res. 2018. PMID: 29112736 Free PMC article.
-
Multi-omics approach dissects cis-regulatory mechanisms underlying North Carolina macular dystrophy, a retinal enhanceropathy.Am J Hum Genet. 2022 Nov 3;109(11):2029-2048. doi: 10.1016/j.ajhg.2022.09.013. Epub 2022 Oct 14. Am J Hum Genet. 2022. PMID: 36243009 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
