

CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain
- PMID: 25999473
- PMCID: PMC4524106
- DOI: 10.1242/jcs.167270
CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain
Abstract
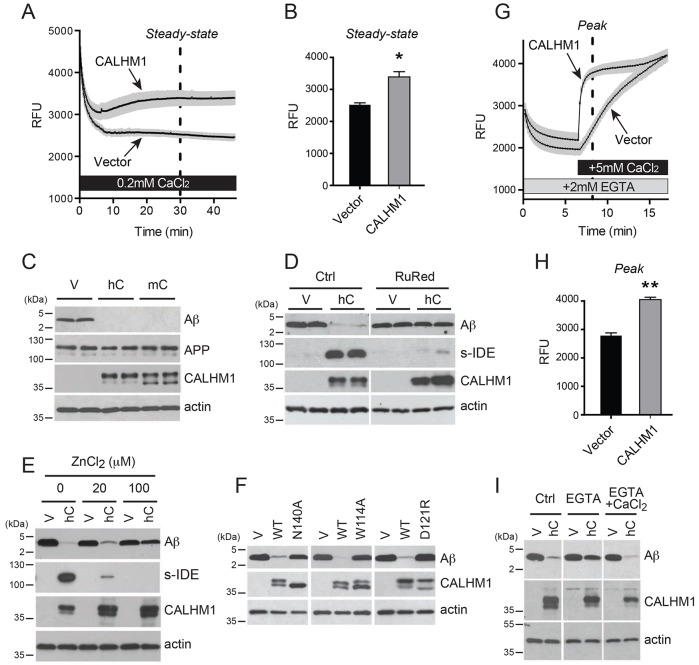
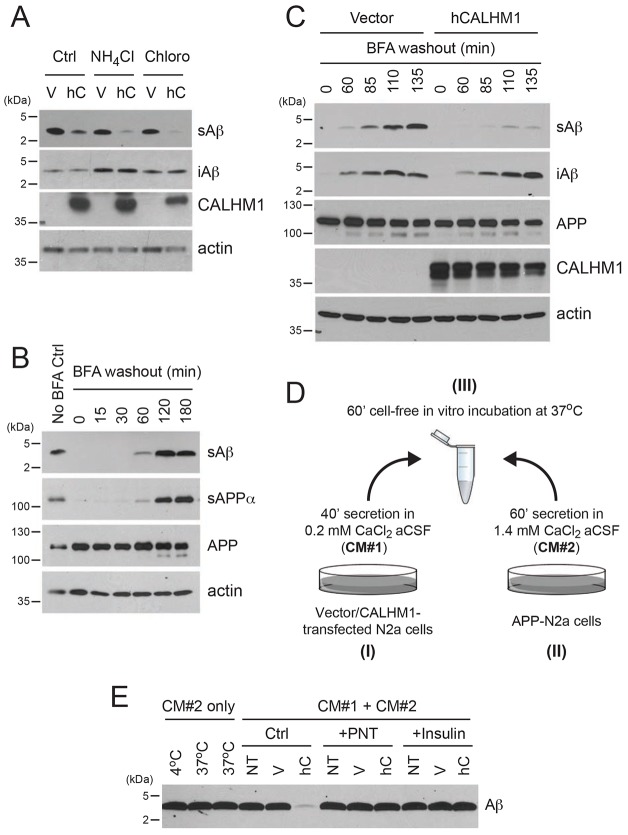
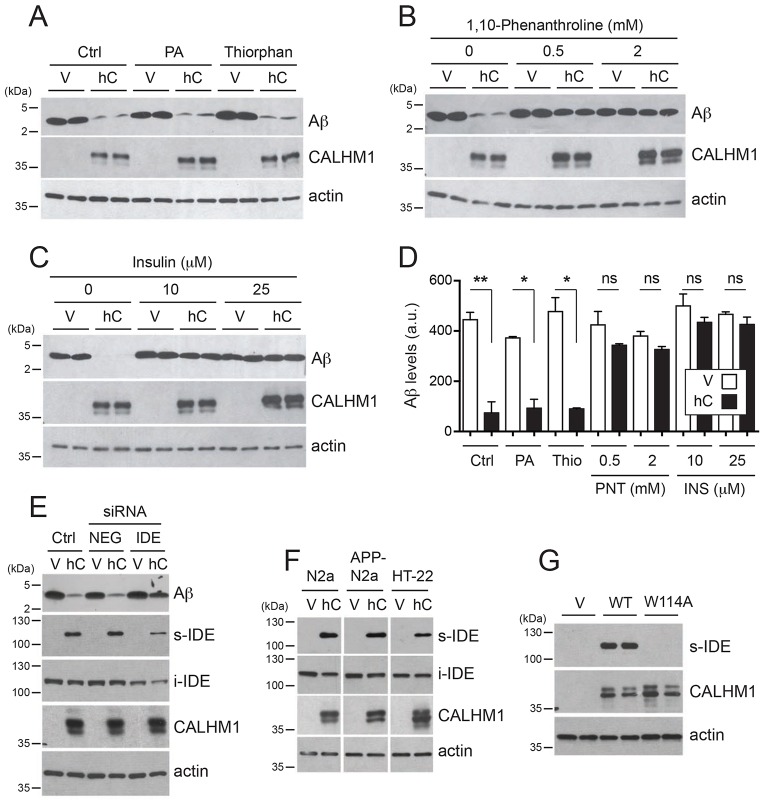
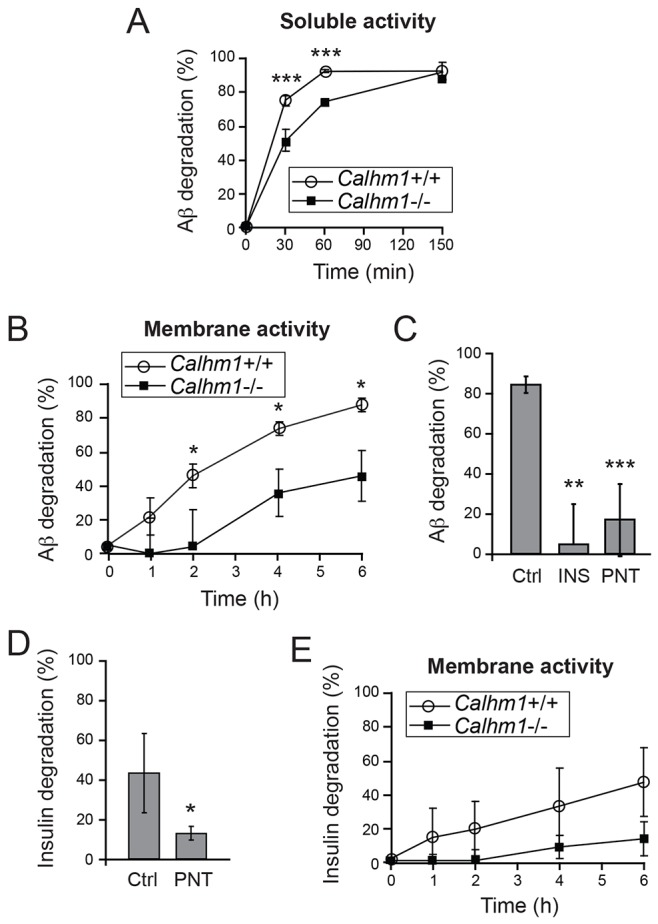
Alzheimer's disease is characterized by amyloid-β (Aβ) peptide accumulation in the brain. CALHM1, a cell-surface Ca(2+) channel expressed in brain neurons, has anti-amyloidogenic properties in cell cultures. Here, we show that CALHM1 controls Aβ levels in vivo in the mouse brain through a previously unrecognized mechanism of regulation of Aβ clearance. Using pharmacological and genetic approaches in cell lines, we found that CALHM1 ion permeability and extracellular Ca(2+) were required for the Aβ-lowering effect of CALHM1. Aβ level reduction by CALHM1 could be explained by an increase in extracellular Aβ degradation by insulin-degrading enzyme (IDE), extracellular secretion of which was strongly potentiated by CALHM1 activation. Importantly, Calhm1 knockout in mice reduced IDE enzymatic activity in the brain, and increased endogenous Aβ concentrations by up to ∼50% in both the whole brain and primary neurons. Thus, CALHM1 controls Aβ levels in cell lines and in vivo by facilitating neuronal and Ca(2+)-dependent degradation of extracellular Aβ by IDE. This work identifies CALHM1 ion channel as a potential target for promoting amyloid clearance in Alzheimer's disease.
Keywords: Alzheimer's disease; Amyloid-β peptide; CALHM1; Insulin-degrading enzyme; Ion channel; Secretion.
© 2015. Published by The Company of Biologists Ltd.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures
Similar articles
-
Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme.J Neurosci. 2000 Mar 1;20(5):1657-65. doi: 10.1523/JNEUROSCI.20-05-01657.2000. J Neurosci. 2000. PMID: 10684867 Free PMC article.
-
CALHM1 and its polymorphism P86L differentially control Ca²⁺homeostasis, mitogen-activated protein kinase signaling, and cell vulnerability upon exposure to amyloid β.Aging Cell. 2015 Dec;14(6):1094-102. doi: 10.1111/acel.12403. Epub 2015 Sep 29. Aging Cell. 2015. PMID: 26416646 Free PMC article.
-
Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo.Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):4162-7. doi: 10.1073/pnas.0230450100. Epub 2003 Mar 12. Proc Natl Acad Sci U S A. 2003. PMID: 12634421 Free PMC article.
-
Powering Amyloid Beta Degrading Enzymes: A Possible Therapy for Alzheimer's Disease.Neurochem Res. 2019 Jun;44(6):1289-1296. doi: 10.1007/s11064-019-02756-x. Epub 2019 Feb 26. Neurochem Res. 2019. PMID: 30806879 Review.
-
Impact of Insulin Degrading Enzyme and Neprilysin in Alzheimer's Disease Biology: Characterization of Putative Cognates for Therapeutic Applications.J Alzheimers Dis. 2015;48(4):891-917. doi: 10.3233/JAD-150379. J Alzheimers Dis. 2015. PMID: 26444774 Review.
Cited by
-
CALHM3 Is Essential for Rapid Ion Channel-Mediated Purinergic Neurotransmission of GPCR-Mediated Tastes.Neuron. 2018 May 2;98(3):547-561.e10. doi: 10.1016/j.neuron.2018.03.043. Epub 2018 Apr 19. Neuron. 2018. PMID: 29681531 Free PMC article.
-
Blockade and knock-out of CALHM1 channels attenuate ischemic brain damage.J Cereb Blood Flow Metab. 2018 Jun;38(6):1060-1069. doi: 10.1177/0271678X17713587. Epub 2017 Jun 9. J Cereb Blood Flow Metab. 2018. PMID: 28597712 Free PMC article.
-
A modification-specific peptide-based immunization approach using CRM197 carrier protein: Development of a selective vaccine against pyroglutamate Aβ peptides.Mol Med. 2017 Jan;22:841-849. doi: 10.2119/molmed.2016.00218. Epub 2016 Nov 28. Mol Med. 2017. PMID: 27900387 Free PMC article.
-
Validation of a priori candidate Alzheimer's disease SNPs with brain amyloid-beta deposition.Sci Rep. 2019 Nov 19;9(1):17069. doi: 10.1038/s41598-019-53604-5. Sci Rep. 2019. PMID: 31745181 Free PMC article.
-
Human CALHM5: Insight in large pore lipid gating ATP channel and associated neurological pathologies.Mol Cell Biochem. 2021 Oct;476(10):3711-3718. doi: 10.1007/s11010-021-04198-y. Epub 2021 Jun 5. Mol Cell Biochem. 2021. PMID: 34089472 Review.
References
-
- Benilova I., Gallardo R., Ungureanu A.-A., Castillo Cano V., Snellinx A., Ramakers M., Bartic C., Rousseau F., Schymkowitz J. and De Strooper B. (2014). The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 289, 30977-30989. 10.1074/jbc.M114.599027 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
