

Remarkably Divergent Regions Punctuate the Genome Assembly of the Caenorhabditis elegans Hawaiian Strain CB4856
- PMID: 25995208
- PMCID: PMC4512556
- DOI: 10.1534/genetics.115.175950
Remarkably Divergent Regions Punctuate the Genome Assembly of the Caenorhabditis elegans Hawaiian Strain CB4856
Abstract
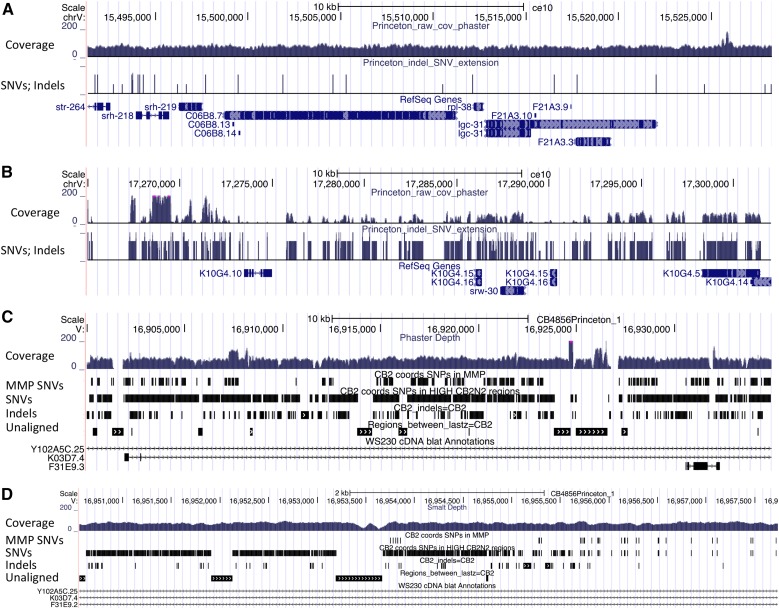
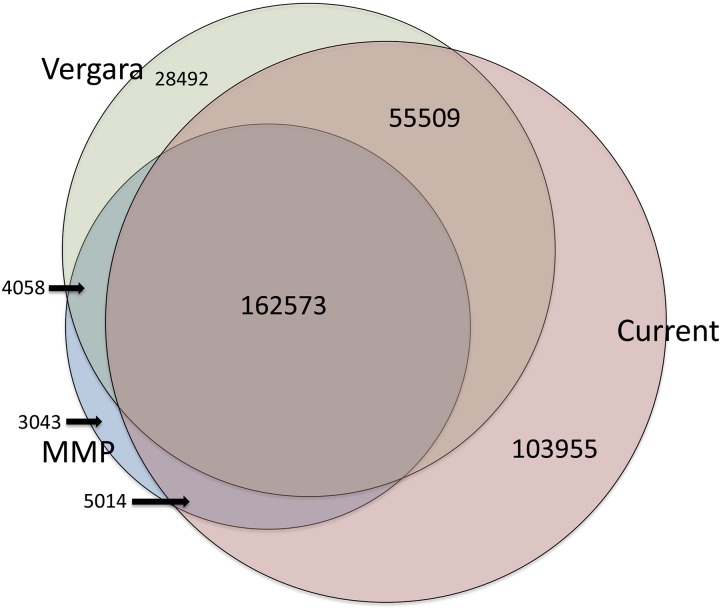
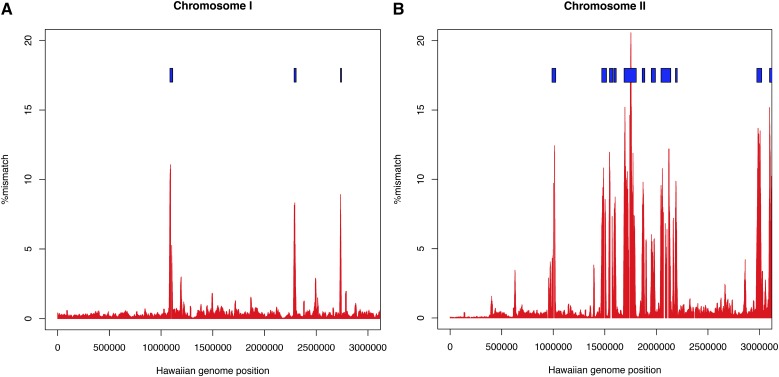
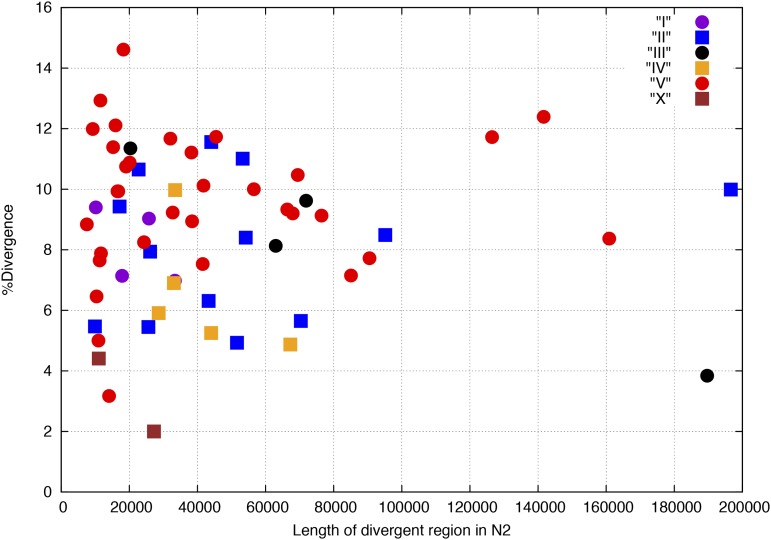
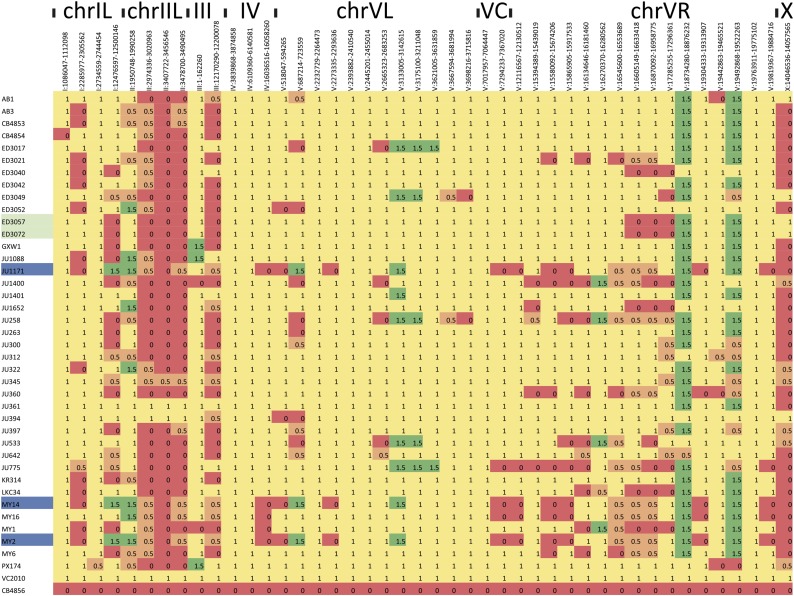
The Hawaiian strain (CB4856) of Caenorhabditis elegans is one of the most divergent from the canonical laboratory strain N2 and has been widely used in developmental, population, and evolutionary studies. To enhance the utility of the strain, we have generated a draft sequence of the CB4856 genome, exploiting a variety of resources and strategies. When compared against the N2 reference, the CB4856 genome has 327,050 single nucleotide variants (SNVs) and 79,529 insertion-deletion events that result in a total of 3.3 Mb of N2 sequence missing from CB4856 and 1.4 Mb of sequence present in CB4856 but not present in N2. As previously reported, the density of SNVs varies along the chromosomes, with the arms of chromosomes showing greater average variation than the centers. In addition, we find 61 regions totaling 2.8 Mb, distributed across all six chromosomes, which have a greatly elevated SNV density, ranging from 2 to 16% SNVs. A survey of other wild isolates show that the two alternative haplotypes for each region are widely distributed, suggesting they have been maintained by balancing selection over long evolutionary times. These divergent regions contain an abundance of genes from large rapidly evolving families encoding F-box, MATH, BATH, seven-transmembrane G-coupled receptors, and nuclear hormone receptors, suggesting that they provide selective advantages in natural environments. The draft sequence makes available a comprehensive catalog of sequence differences between the CB4856 and N2 strains that will facilitate the molecular dissection of their phenotypic differences. Our work also emphasizes the importance of going beyond simple alignment of reads to a reference genome when assessing differences between genomes.
Keywords: C. elegans; evolution; genome assembly; variation.
Copyright © 2015 by the Genetics Society of America.
Figures

Similar articles
-
Punctuated Loci on Chromosome IV Determine Natural Variation in Orsay Virus Susceptibility of Caenorhabditis elegans Strains Bristol N2 and Hawaiian CB4856.J Virol. 2021 May 24;95(12):e02430-20. doi: 10.1128/JVI.02430-20. Print 2021 May 24. J Virol. 2021. PMID: 33827942 Free PMC article.
-
Genome-wide variations in a natural isolate of the nematode Caenorhabditis elegans.BMC Genomics. 2014 Apr 2;15:255. doi: 10.1186/1471-2164-15-255. BMC Genomics. 2014. PMID: 24694239 Free PMC article.
-
Shifting patterns of natural variation in the nuclear genome of caenorhabditis elegans.BMC Evol Biol. 2011 Jun 16;11:168. doi: 10.1186/1471-2148-11-168. BMC Evol Biol. 2011. PMID: 21679441 Free PMC article.
-
Genome evolution in Caenorhabditis.Brief Funct Genomic Proteomic. 2008 May;7(3):211-6. doi: 10.1093/bfgp/eln022. Epub 2008 Jun 23. Brief Funct Genomic Proteomic. 2008. PMID: 18573804 Review.
-
The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans.Genome Biol. 2003;4(12):238. doi: 10.1186/gb-2003-4-12-238. Epub 2003 Nov 18. Genome Biol. 2003. PMID: 14659008 Free PMC article. Review.
Cited by
-
Mutation Is a Sufficient and Robust Predictor of Genetic Variation for Mitotic Spindle Traits in Caenorhabditis elegans.Genetics. 2016 Aug;203(4):1859-70. doi: 10.1534/genetics.115.185736. Epub 2016 Jun 22. Genetics. 2016. PMID: 27334268 Free PMC article.
-
PhenoMIP: High-Throughput Phenotyping of Diverse Caenorhabditis elegans Populations via Molecular Inversion Probes.G3 (Bethesda). 2020 Nov 5;10(11):3977-3990. doi: 10.1534/g3.120.401656. G3 (Bethesda). 2020. PMID: 32868407 Free PMC article.
-
Fine-Scale Crossover Rate Variation on the Caenorhabditis elegans X Chromosome.G3 (Bethesda). 2016 Jun 1;6(6):1767-76. doi: 10.1534/g3.116.028001. G3 (Bethesda). 2016. PMID: 27172189 Free PMC article.
-
Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions.Nat Commun. 2019 Apr 12;10(1):1702. doi: 10.1038/s41467-019-09575-2. Nat Commun. 2019. PMID: 30979905 Free PMC article.
-
The zinc transporter ZIPT-7.1 regulates sperm activation in nematodes.PLoS Biol. 2018 Jun 7;16(6):e2005069. doi: 10.1371/journal.pbio.2005069. eCollection 2018 Jun. PLoS Biol. 2018. PMID: 29879108 Free PMC article.
References
-
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- SRA/SRX1001806
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
