

Live to die another way: modes of programmed cell death and the signals emanating from dying cells
- PMID: 25991373
- PMCID: PMC4511109
- DOI: 10.1038/nrm3999
Live to die another way: modes of programmed cell death and the signals emanating from dying cells
Abstract
All life ends in death, but perhaps one of life's grander ironies is that it also depends on death. Cell-intrinsic suicide pathways, termed programmed cell death (PCD), are crucial for animal development, tissue homeostasis and pathogenesis. Originally, PCD was almost synonymous with apoptosis; recently, however, alternative mechanisms of PCD have been reported. Here, we provide an overview of several distinct PCD mechanisms, namely apoptosis, autophagy and necroptosis. In addition, we discuss the complex signals that emanate from dying cells, which can either trigger regeneration or instruct additional killing. Further advances in understanding the physiological roles of the various mechanisms of cell death and their associated signals will be important to selectively manipulate PCD for therapeutic purposes.
Conflict of interest statement
The authors declare no competing interests.
Figures
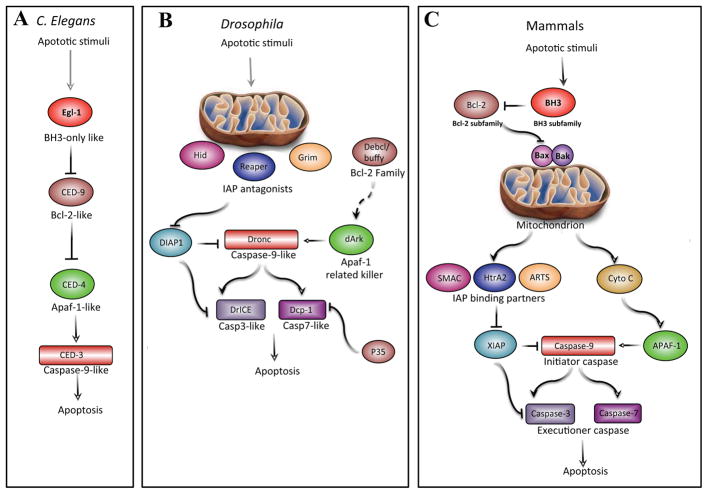
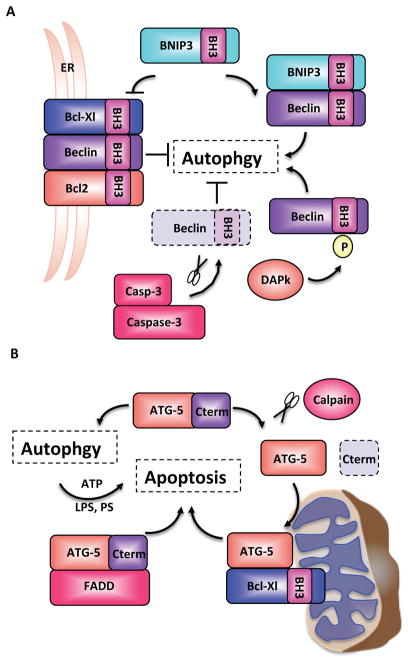
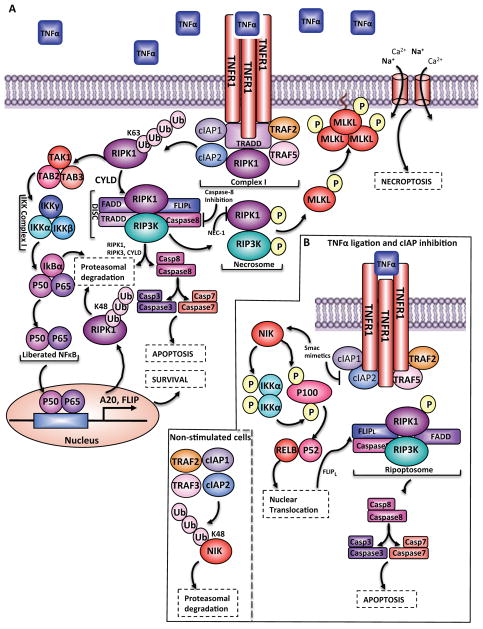
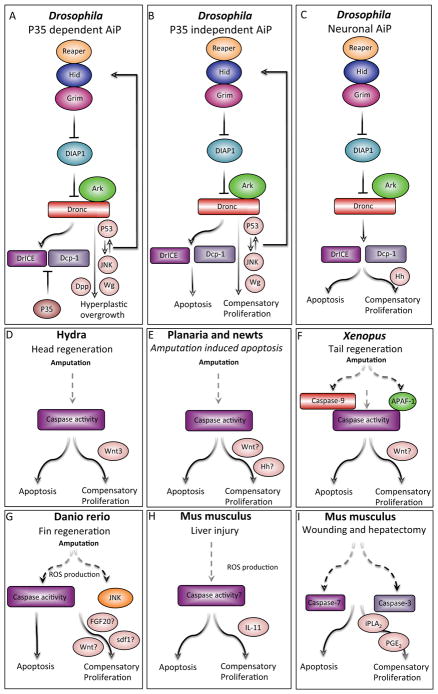
Similar articles
-
Another way to die: autophagic programmed cell death.Cell Death Differ. 2005 Nov;12 Suppl 2:1528-34. doi: 10.1038/sj.cdd.4401777. Cell Death Differ. 2005. PMID: 16247500 Review.
-
Mechanical insights into the regulation of programmed cell death by p53 via mitochondria.Biochim Biophys Acta Mol Cell Res. 2019 May;1866(5):839-848. doi: 10.1016/j.bbamcr.2019.02.009. Epub 2019 Feb 18. Biochim Biophys Acta Mol Cell Res. 2019. PMID: 30790591 Review.
-
Resilience of death: intrinsic disorder in proteins involved in the programmed cell death.Cell Death Differ. 2013 Sep;20(9):1257-67. doi: 10.1038/cdd.2013.65. Epub 2013 Jun 14. Cell Death Differ. 2013. PMID: 23764774 Free PMC article.
-
Programmed cell death in tumor immunity: mechanistic insights and clinical implications.Front Immunol. 2024 Jan 12;14:1309635. doi: 10.3389/fimmu.2023.1309635. eCollection 2023. Front Immunol. 2024. PMID: 38283351 Free PMC article. Review.
-
Emerging connectivity of programmed cell death pathways and its physiological implications.Nat Rev Mol Cell Biol. 2020 Nov;21(11):678-695. doi: 10.1038/s41580-020-0270-8. Epub 2020 Sep 1. Nat Rev Mol Cell Biol. 2020. PMID: 32873928 Review.
Cited by
-
CMLS forum reviews: mitochondrial damage control.Cell Mol Life Sci. 2021 Apr;78(8):3763-3765. doi: 10.1007/s00018-021-03804-y. Epub 2021 Mar 12. Cell Mol Life Sci. 2021. PMID: 33710354 Free PMC article.
-
Cytokine regulation of apoptosis-induced apoptosis and apoptosis-induced cell proliferation in vascular smooth muscle cells.Apoptosis. 2020 Oct;25(9-10):648-662. doi: 10.1007/s10495-020-01622-4. Apoptosis. 2020. PMID: 32627119 Free PMC article.
-
Machine learning-based cell death marker for predicting prognosis and identifying tumor immune microenvironment in prostate cancer.Heliyon. 2024 Sep 6;10(18):e37554. doi: 10.1016/j.heliyon.2024.e37554. eCollection 2024 Sep 30. Heliyon. 2024. PMID: 39309810 Free PMC article.
-
Identification and verification of disulfidptosis-related genes in sepsis-induced acute lung injury.Front Med (Lausanne). 2024 Aug 28;11:1430252. doi: 10.3389/fmed.2024.1430252. eCollection 2024. Front Med (Lausanne). 2024. PMID: 39262873 Free PMC article.
-
Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes.Cells. 2021 May 20;10(5):1265. doi: 10.3390/cells10051265. Cells. 2021. PMID: 34065321 Free PMC article. Review.
References
-
- Vogt C. Untersuchungen über die Entwicklungsgeschichte der Geburtshelferkröte (Alytes obstetricans) Solothurn: Jent und Gassman. 1842:130.
-
- Lockshin RA, Williams CM. Programmed cell death. II Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol Dev. 1964;10:643–64.
-
- Tata JR. Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Dev Biol. 1966;13:77–94. - PubMed
-
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
