

Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility
- PMID: 25984238
- PMCID: PMC4432502
- DOI: 10.1186/s13072-015-0009-5
Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility
Abstract
Background: Regulated gene expression controls organismal development, and variation in regulatory patterns has been implicated in complex traits. Thus accurate prediction of enhancers is important for further understanding of these processes. Genome-wide measurement of epigenetic features, such as histone modifications and occupancy by transcription factors, is improving enhancer predictions, but the contribution of these features to prediction accuracy is not known. Given the importance of the hematopoietic transcription factor TAL1 for erythroid gene activation, we predicted candidate enhancers based on genomic occupancy by TAL1 and measured their activity. Contributions of multiple features to enhancer prediction were evaluated based on the results of these and other studies.
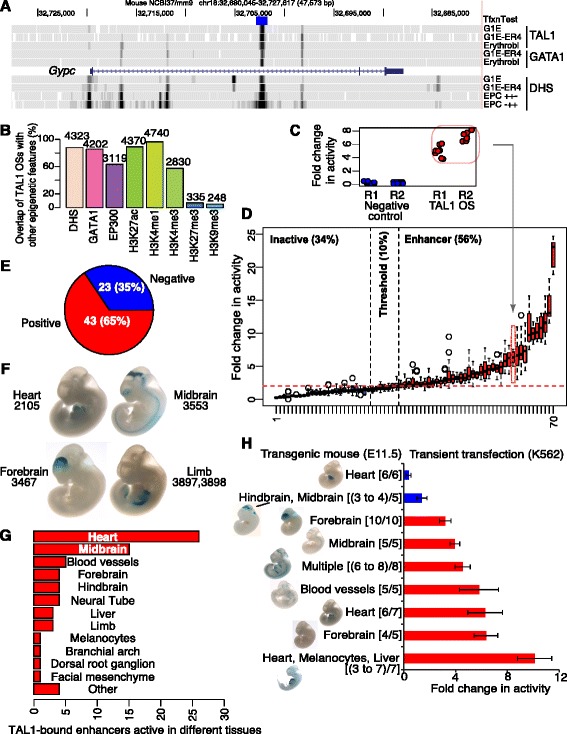
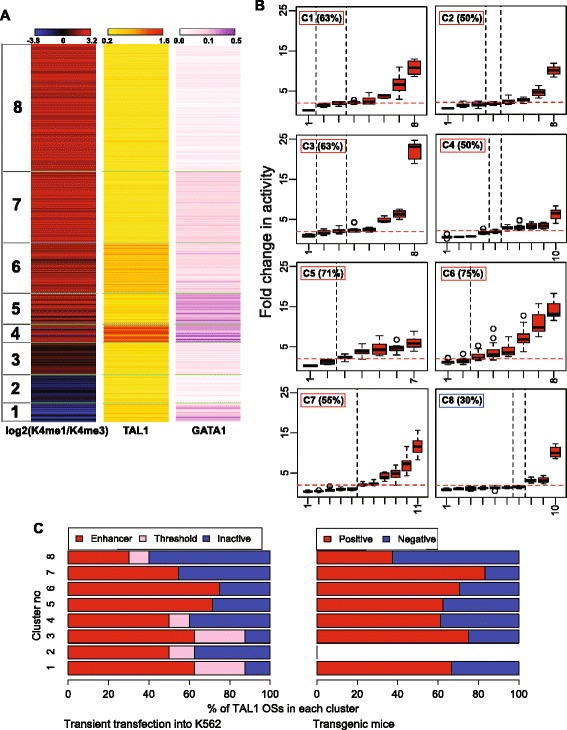
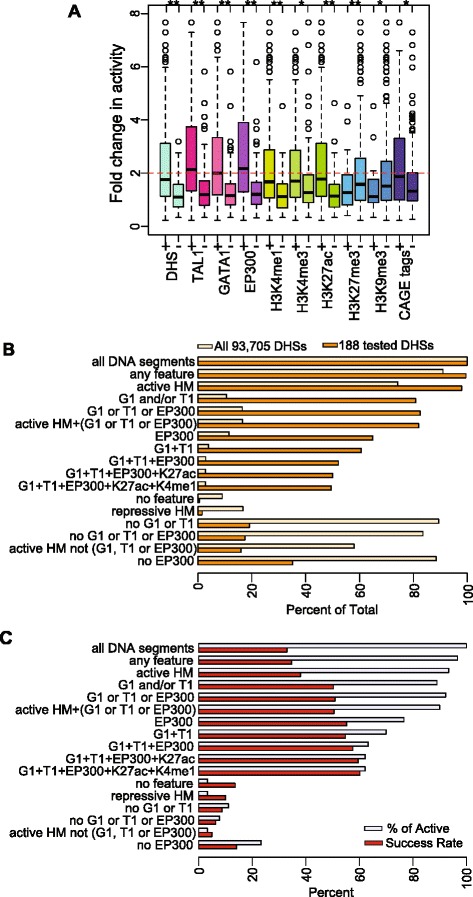
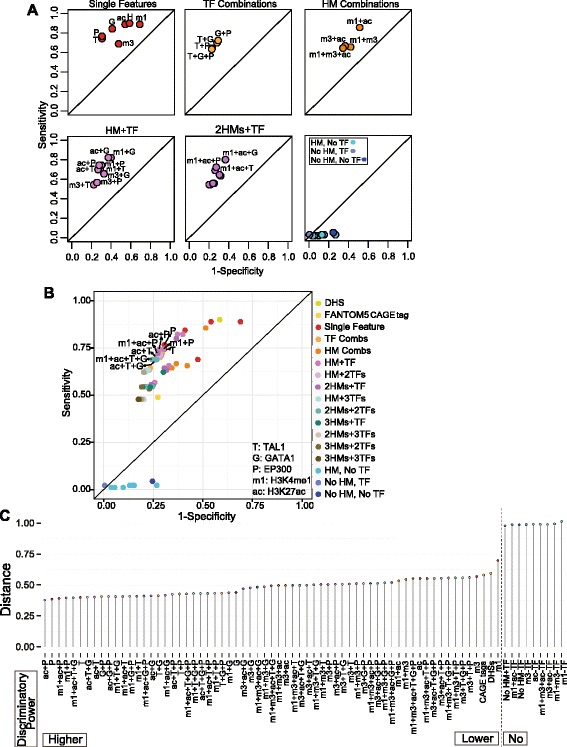
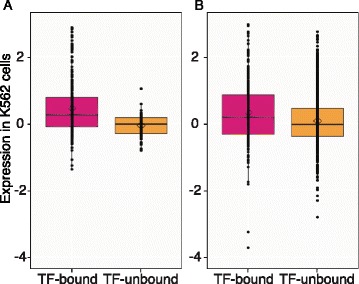
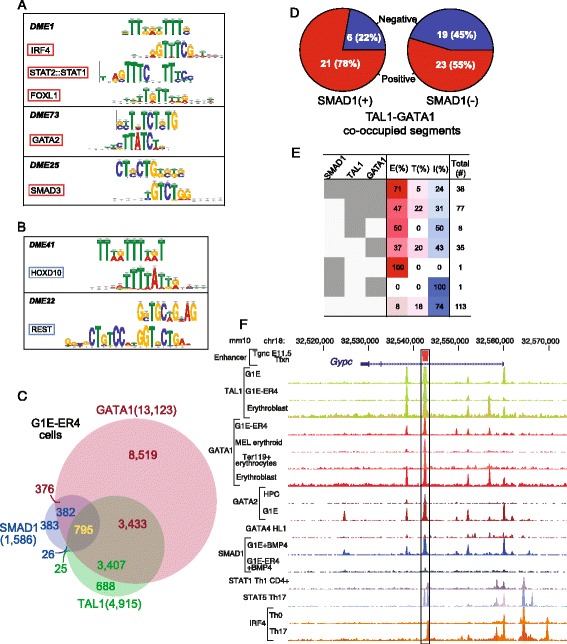
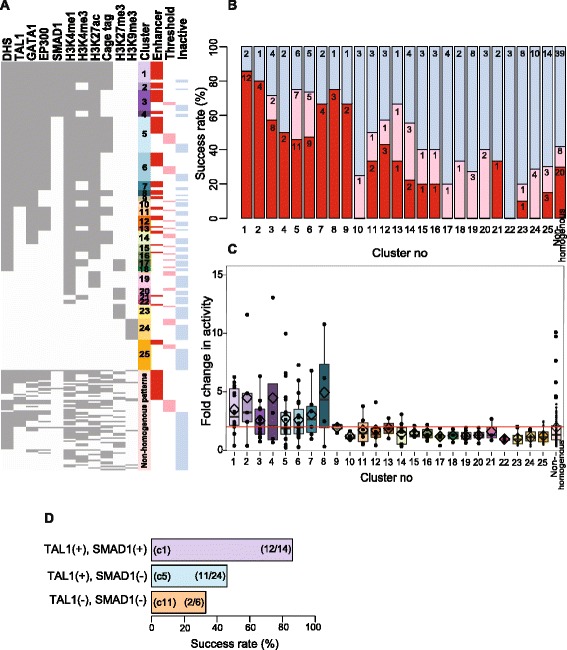
Results: TAL1-bound DNA segments were active enhancers at a high rate both in transient transfections of cultured cells (39 of 79, or 56%) and transgenic mice (43 of 66, or 65%). The level of binding signal for TAL1 or GATA1 did not help distinguish TAL1-bound DNA segments as active versus inactive enhancers, nor did the density of regulation-related histone modifications. A meta-analysis of results from this and other studies (273 tested predicted enhancers) showed that the presence of TAL1, GATA1, EP300, SMAD1, H3K4 methylation, H3K27ac, and CAGE tags at DNase hypersensitive sites gave the most accurate predictors of enhancer activity, with a success rate over 80% and a median threefold increase in activity. Chromatin accessibility assays and the histone modifications H3K4me1 and H3K27ac were sensitive for finding enhancers, but they have high false positive rates unless transcription factor occupancy is also included.
Conclusions: Occupancy by key transcription factors such as TAL1, GATA1, SMAD1, and EP300, along with evidence of transcription, improves the accuracy of enhancer predictions based on epigenetic features.
Keywords: Enhancer assay; Functional genomics; GATA1; Gene regulation; Histone modifications; TAL1.
Figures
Similar articles
-
Hematopoietic/erythroid enhancers activate nearby target genes by extending histone H3K27ac and transcribing intergenic RNA.FASEB J. 2023 Apr;37(4):e22870. doi: 10.1096/fj.202201891R. FASEB J. 2023. PMID: 36929052
-
Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression.Genome Res. 2009 Dec;19(12):2172-84. doi: 10.1101/gr.098921.109. Epub 2009 Nov 3. Genome Res. 2009. PMID: 19887574 Free PMC article.
-
Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers.Epigenetics Chromatin. 2016 Sep 9;9(1):37. doi: 10.1186/s13072-016-0087-z. eCollection 2016. Epigenetics Chromatin. 2016. PMID: 27617035 Free PMC article.
-
Epigenetic plasticity of enhancers in cancer.Transcription. 2020 Feb;11(1):26-36. doi: 10.1080/21541264.2020.1713682. Epub 2020 Jan 16. Transcription. 2020. PMID: 31944157 Free PMC article. Review.
-
A survey of recently emerged genome-wide computational enhancer predictor tools.Comput Biol Chem. 2018 Jun;74:132-141. doi: 10.1016/j.compbiolchem.2018.03.019. Epub 2018 Mar 16. Comput Biol Chem. 2018. PMID: 29602043 Review.
Cited by
-
Single-nucleotide-level mapping of DNA regulatory elements that control fetal hemoglobin expression.Nat Genet. 2021 Jun;53(6):869-880. doi: 10.1038/s41588-021-00861-8. Epub 2021 May 6. Nat Genet. 2021. PMID: 33958780 Free PMC article.
-
A map of cis-regulatory modules and constituent transcription factor binding sites in 80% of the mouse genome.BMC Genomics. 2022 Oct 19;23(1):714. doi: 10.1186/s12864-022-08933-7. BMC Genomics. 2022. PMID: 36261804 Free PMC article.
-
Repurposing of promoters and enhancers during mammalian evolution.Nat Commun. 2018 Oct 4;9(1):4066. doi: 10.1038/s41467-018-06544-z. Nat Commun. 2018. PMID: 30287902 Free PMC article.
-
Concise Review: Epigenetic Regulation of Hematopoiesis: Biological Insights and Therapeutic Applications.Stem Cells Transl Med. 2017 Dec;6(12):2106-2114. doi: 10.1002/sctm.17-0192. Epub 2017 Oct 28. Stem Cells Transl Med. 2017. PMID: 29080249 Free PMC article. Review.
-
Multiple enhancer regions govern the transcription of CCN2 during embryonic development.J Cell Commun Signal. 2018 Mar;12(1):231-243. doi: 10.1007/s12079-017-0440-4. Epub 2017 Dec 18. J Cell Commun Signal. 2018. PMID: 29256171 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
