

Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast
- PMID: 25950722
- PMCID: PMC4423881
- DOI: 10.1371/journal.pgen.1005206
Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast
Abstract
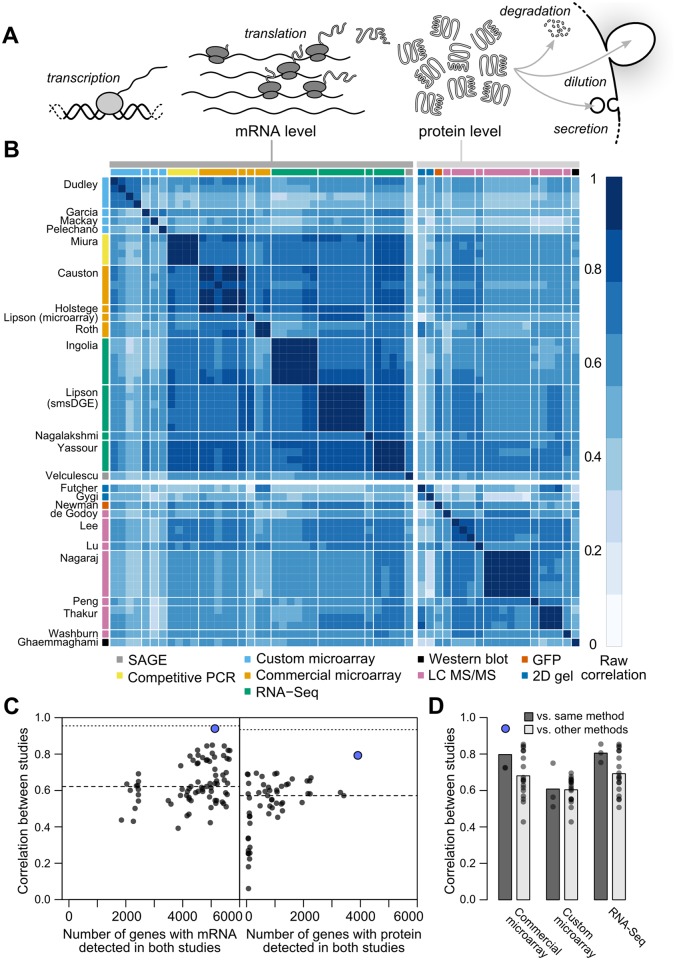
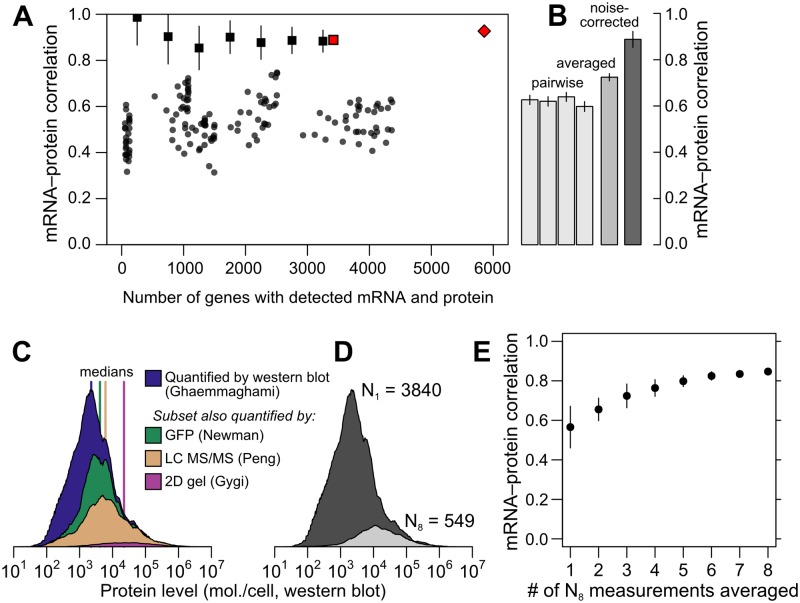
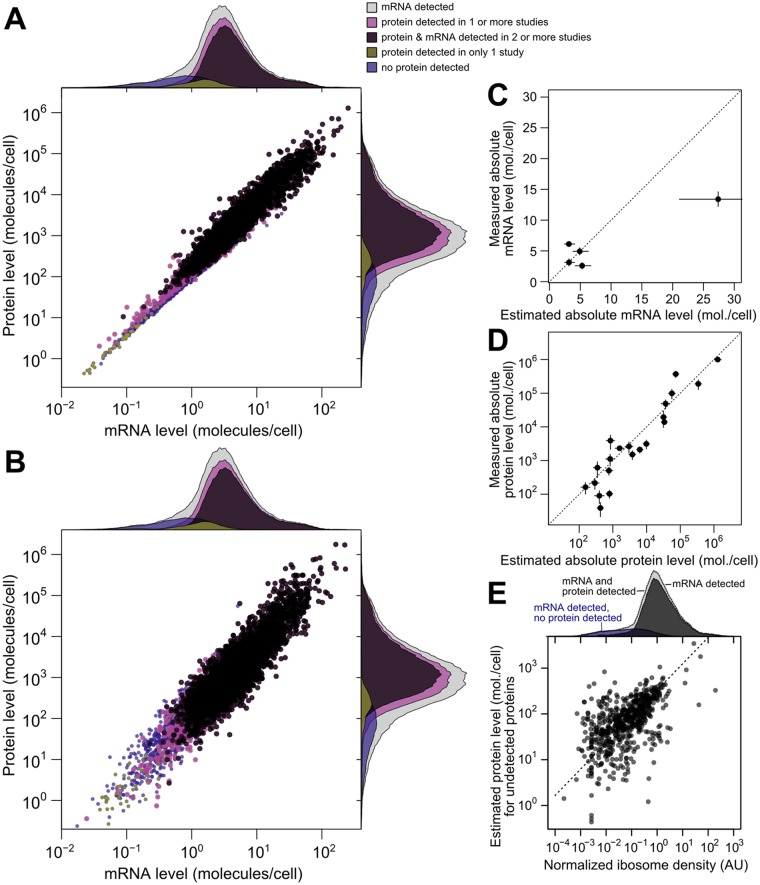
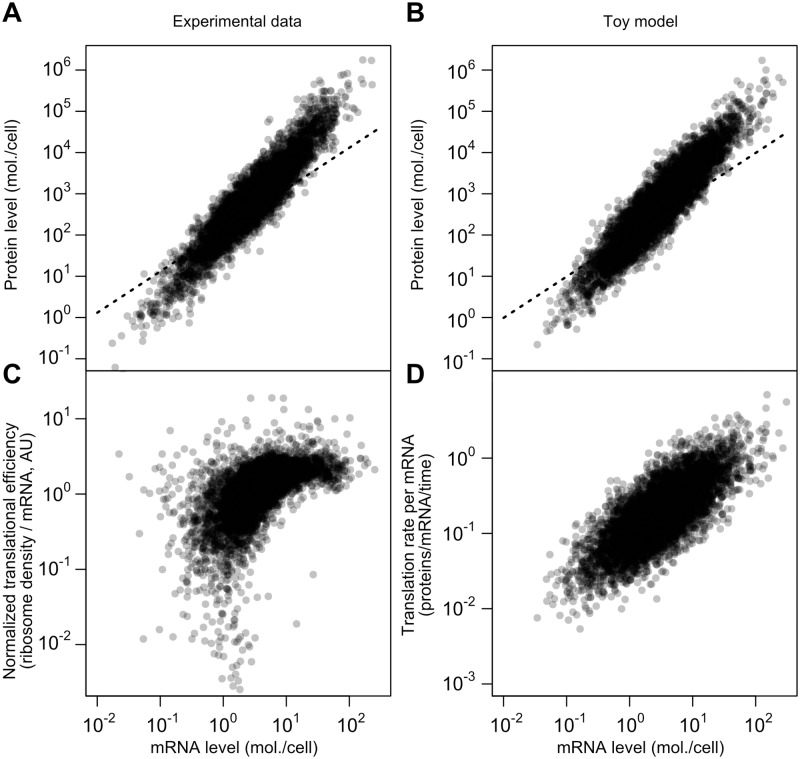
Cells respond to their environment by modulating protein levels through mRNA transcription and post-transcriptional control. Modest observed correlations between global steady-state mRNA and protein measurements have been interpreted as evidence that mRNA levels determine roughly 40% of the variation in protein levels, indicating dominant post-transcriptional effects. However, the techniques underlying these conclusions, such as correlation and regression, yield biased results when data are noisy, missing systematically, and collinear---properties of mRNA and protein measurements---which motivated us to revisit this subject. Noise-robust analyses of 24 studies of budding yeast reveal that mRNA levels explain more than 85% of the variation in steady-state protein levels. Protein levels are not proportional to mRNA levels, but rise much more rapidly. Regulation of translation suffices to explain this nonlinear effect, revealing post-transcriptional amplification of, rather than competition with, transcriptional signals. These results substantially revise widely credited models of protein-level regulation, and introduce multiple noise-aware approaches essential for proper analysis of many biological phenomena.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
Post-transcriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale.Mol Cell Proteomics. 2004 Nov;3(11):1083-92. doi: 10.1074/mcp.M400099-MCP200. Epub 2004 Aug 23. Mol Cell Proteomics. 2004. PMID: 15326222
-
Post-transcriptional regulation in the myo1Δ mutant of Saccharomyces cerevisiae.BMC Genomics. 2010 Dec 2;11:690. doi: 10.1186/1471-2164-11-690. BMC Genomics. 2010. PMID: 21126371 Free PMC article.
-
Spatio-temporal dynamics of yeast mitochondrial biogenesis: transcriptional and post-transcriptional mRNA oscillatory modules.PLoS Comput Biol. 2009 Jun;5(6):e1000409. doi: 10.1371/journal.pcbi.1000409. Epub 2009 Jun 12. PLoS Comput Biol. 2009. PMID: 19521515 Free PMC article.
-
Post-transcriptional regulation in budding yeast meiosis.Curr Genet. 2016 May;62(2):313-5. doi: 10.1007/s00294-015-0546-2. Epub 2015 Nov 27. Curr Genet. 2016. PMID: 26613728 Free PMC article. Review.
-
From transcription to mRNA: PAF provides a new path.Mol Cell. 2005 Oct 28;20(2):167-8. doi: 10.1016/j.molcel.2005.10.004. Mol Cell. 2005. PMID: 16246718 Review.
Cited by
-
A dynamical stochastic model of yeast translation across the cell cycle.Heliyon. 2023 Jan 26;9(2):e13101. doi: 10.1016/j.heliyon.2023.e13101. eCollection 2023 Feb. Heliyon. 2023. PMID: 36793957 Free PMC article.
-
Uncovering extensive post-translation regulation during human cell cycle progression by integrative multi-'omics analysis.BMC Bioinformatics. 2019 Oct 29;20(1):536. doi: 10.1186/s12859-019-3150-5. BMC Bioinformatics. 2019. PMID: 31664894 Free PMC article.
-
A deep proteome and transcriptome abundance atlas of 29 healthy human tissues.Mol Syst Biol. 2019 Feb 18;15(2):e8503. doi: 10.15252/msb.20188503. Mol Syst Biol. 2019. PMID: 30777892 Free PMC article.
-
Mass spectrometry-based draft of the mouse proteome.Nat Methods. 2022 Jul;19(7):803-811. doi: 10.1038/s41592-022-01526-y. Epub 2022 Jun 16. Nat Methods. 2022. PMID: 35710609 Free PMC article.
-
Integrated omics dissection of proteome dynamics during cardiac remodeling.Nat Commun. 2018 Jan 9;9(1):120. doi: 10.1038/s41467-017-02467-3. Nat Commun. 2018. PMID: 29317621 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
