

Inside-out Regulation of Ectodomain Cleavage of Cluster-of-Differentiation-44 (CD44) and of Neuregulin-1 Requires Substrate Dimerization
- PMID: 25925953
- PMCID: PMC4498042
- DOI: 10.1074/jbc.M114.610204
Inside-out Regulation of Ectodomain Cleavage of Cluster-of-Differentiation-44 (CD44) and of Neuregulin-1 Requires Substrate Dimerization
Abstract
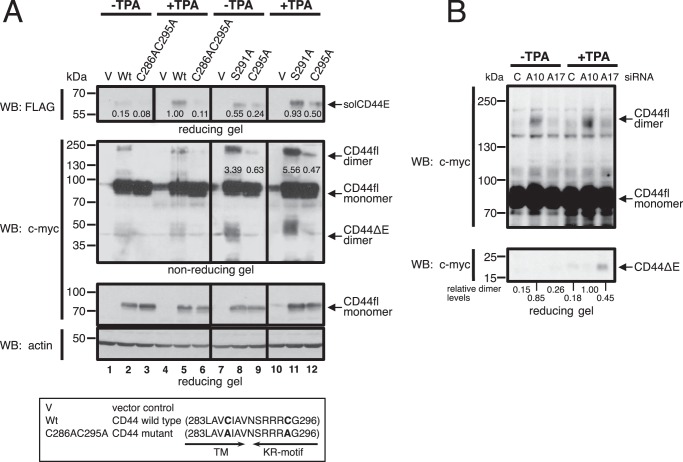
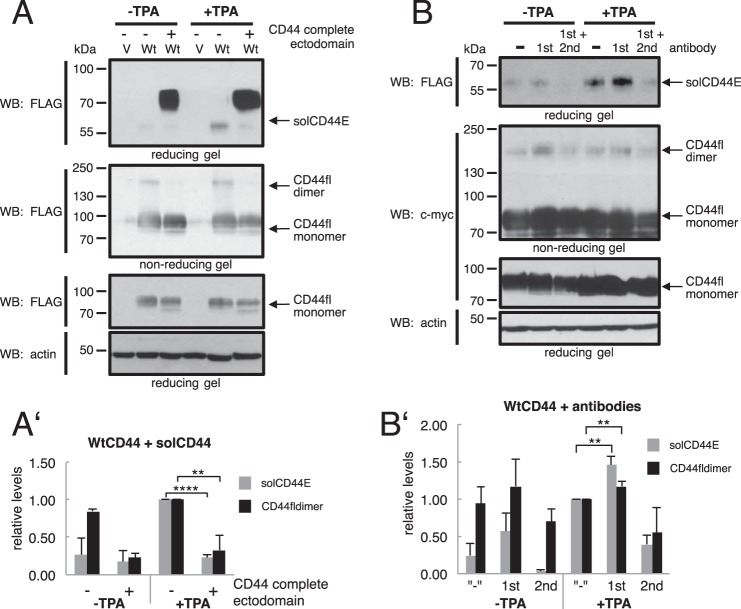
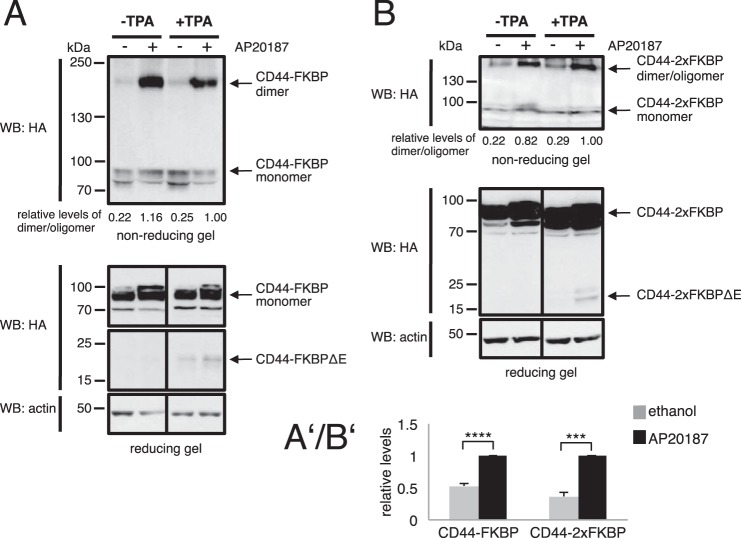
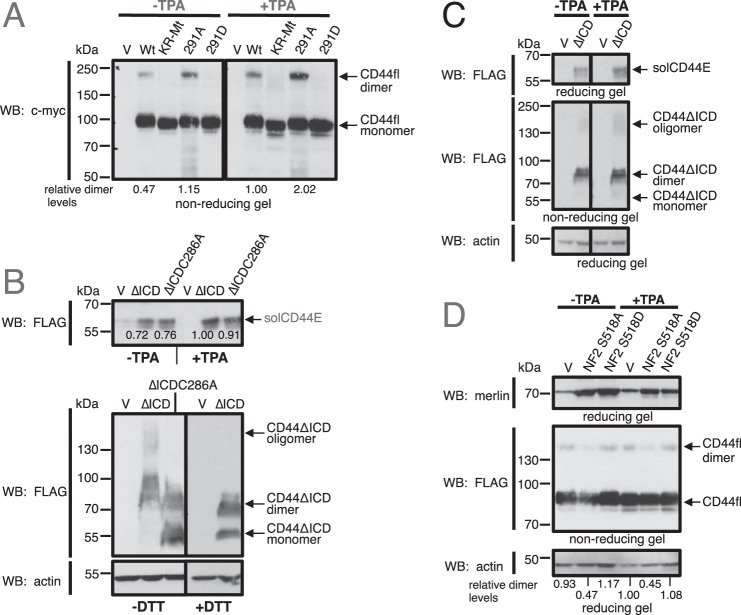
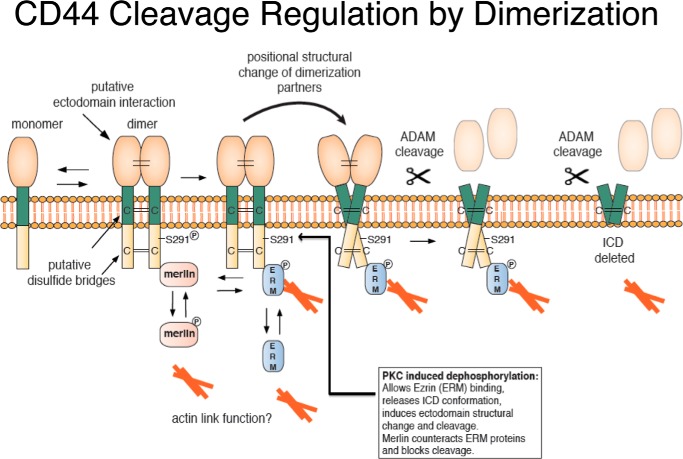
Ectodomain shedding of transmembrane precursor proteins generates numerous life-essential molecules, such as epidermal growth factor receptor ligands. This cleavage not only releases the regulatory growth factor, but it is also the required first step for the subsequent processing by γ-secretase and the release of gene regulatory intracellular fragments. Signaling within the cell modifies the cytoplasmic tails of substrates, a step important in starting the specific and regulated cleavage of a large number of studied substrates. Ectodomain cleavage occurs, however, on the outside of the plasma membrane and is carried out by membrane-bound metalloproteases. How the intracellular domain modification communicates with the ectodomain of the substrate to allow for cleavage to occur is unknown. Here, we show that homodimerization of a cluster-of-differentiation-44 or of pro-neuregulin-1 monomers represents an essential pre-condition for their regulated ectodomain cleavage. Both substrates are associated with their respective metalloproteases under both basal or cleavage-stimulated conditions. These interactions only turn productive by specific intracellular signal-induced intracellular domain modifications of the substrates, which in turn regulate metalloprotease access to the substrates' ectodomain and cleavage. We propose that substrate intracellular domain modification induces a relative rotation or other positional change of the dimerization partners that allow metalloprotease cleavage in the extracellular space. Our findings fill an important gap in understanding substrate-specific inside-out signal transfer along cleaved transmembrane proteins and suggest that substrate dimerization (homo- or possibly heterodimerization) might represent a general principle in ectodomain shedding.
Keywords: ADAM; ADAM10; ADAM17; actin; adhesion molecule; angiotensin; ezrin; metalloprotease; neuregulin.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Growth factor and co-receptor release by structural regulation of substrate metalloprotease accessibility.Sci Rep. 2016 Nov 23;6:37464. doi: 10.1038/srep37464. Sci Rep. 2016. PMID: 27876763 Free PMC article.
-
The Coxsackievirus and Adenovirus Receptor (CAR) undergoes ectodomain shedding and regulated intramembrane proteolysis (RIP).PLoS One. 2013 Aug 28;8(8):e73296. doi: 10.1371/journal.pone.0073296. eCollection 2013. PLoS One. 2013. PMID: 24015300 Free PMC article.
-
Distinct Intracellular Domain Substrate Modifications Selectively Regulate Ectodomain Cleavage of NRG1 or CD44.Mol Cell Biol. 2015 Oct;35(19):3381-95. doi: 10.1128/MCB.00500-15. Epub 2015 Jul 27. Mol Cell Biol. 2015. PMID: 26217011 Free PMC article.
-
Part-time alpha-secretases: the functional biology of ADAM 9, 10 and 17.Curr Alzheimer Res. 2008 Apr;5(2):187-201. doi: 10.2174/156720508783954686. Curr Alzheimer Res. 2008. PMID: 18393804 Review.
-
The "A Disintegrin And Metalloproteases" ADAM10 and ADAM17: novel drug targets with therapeutic potential?Eur J Cell Biol. 2011 Jun-Jul;90(6-7):527-35. doi: 10.1016/j.ejcb.2010.11.005. Epub 2010 Dec 30. Eur J Cell Biol. 2011. PMID: 21194787 Review.
Cited by
-
Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies.Sci Adv. 2021 Nov 26;7(48):eabj1826. doi: 10.1126/sciadv.abj1826. Epub 2021 Nov 24. Sci Adv. 2021. PMID: 34818048 Free PMC article.
-
L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing.Sci Rep. 2019 Mar 6;9(1):3716. doi: 10.1038/s41598-019-39884-x. Sci Rep. 2019. PMID: 30842511 Free PMC article.
-
Activity dependent CAM cleavage and neurotransmission.Front Cell Neurosci. 2015 Aug 11;9:305. doi: 10.3389/fncel.2015.00305. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26321910 Free PMC article. Review.
-
Neuregulin 1: an intriguing therapeutic target for neurodevelopmental disorders.Transl Psychiatry. 2020 Jun 16;10(1):190. doi: 10.1038/s41398-020-00868-5. Transl Psychiatry. 2020. PMID: 32546684 Free PMC article. Review.
-
Gamma secretase dependent release of the CD44 cytoplasmic tail upregulates IFI16 in cd44-/- tumor cells, MEFs and macrophages.PLoS One. 2018 Dec 12;13(12):e0207358. doi: 10.1371/journal.pone.0207358. eCollection 2018. PLoS One. 2018. PMID: 30540779 Free PMC article.
References
-
- van der Vorst E. P., Keijbeck A. A., de Winther M. P., Donners M. M. (2012) A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis 224, 302–308 - PubMed
-
- Klein T., Bischoff R. (2011) Active metalloproteases of the a disintegrin and metalloprotease (ADAM) family: biological function and structure. J. Proteome Res. 10, 17–33 - PubMed
-
- Hartmann M., Herrlich A., Herrlich P. (2013) Who decides when to cleave an ectodomain? Trends Biochem. Sci. 38, 111–120 - PubMed
-
- Dang M., Dubbin K., D'Aiello A., Hartmann M., Lodish H., Herrlich A. (2011) Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. J. Biol. Chem. 286, 17704–17713 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
