

Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity
- PMID: 25919952
- PMCID: PMC4408529
- DOI: 10.7554/eLife.06416
Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity
Abstract
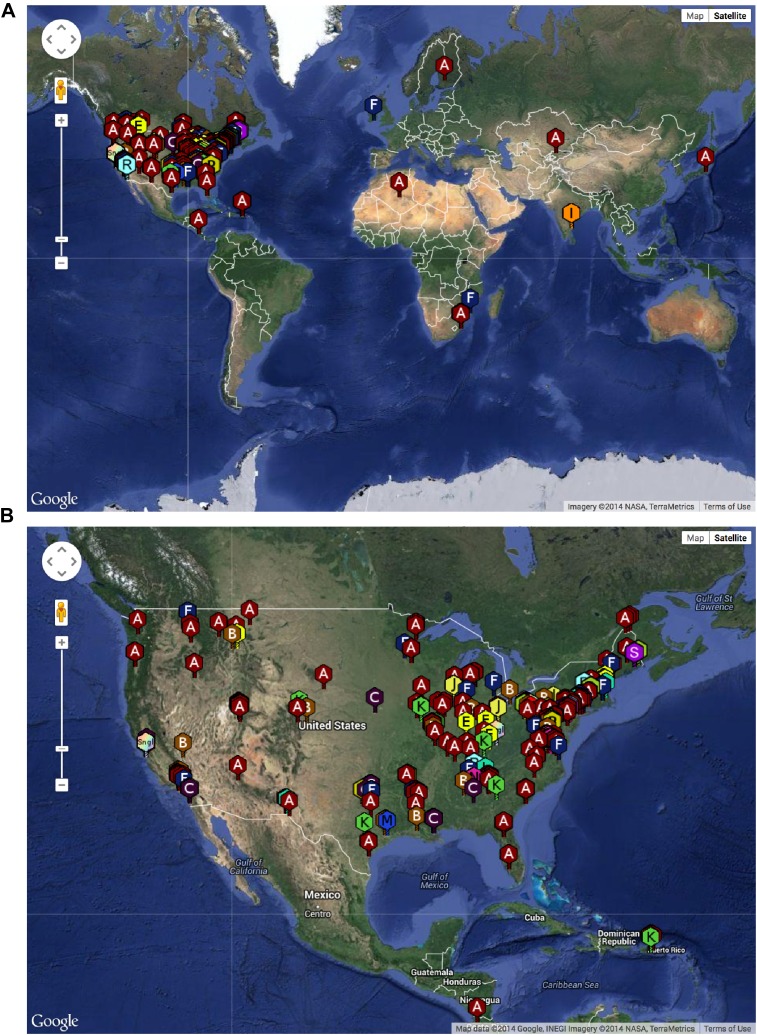
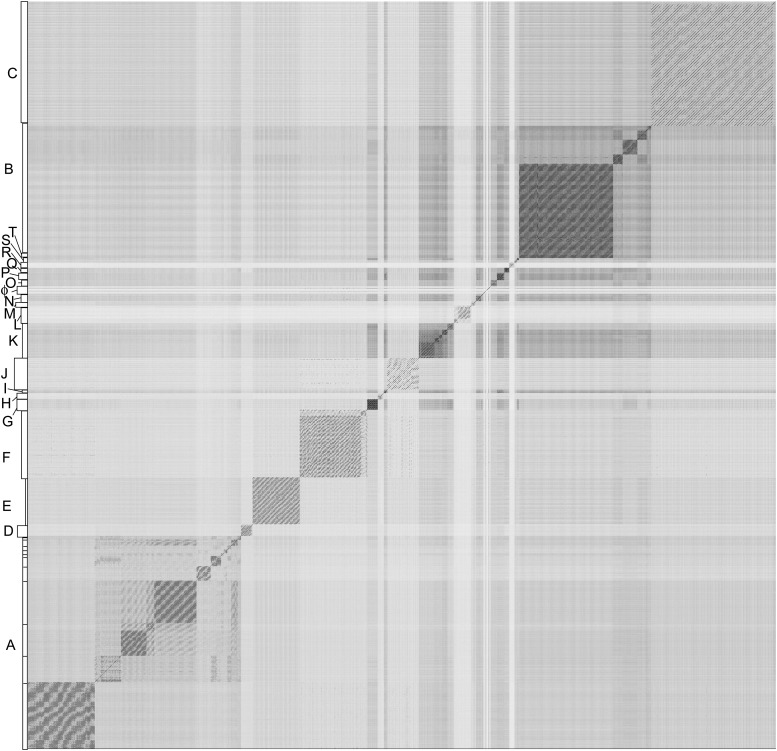
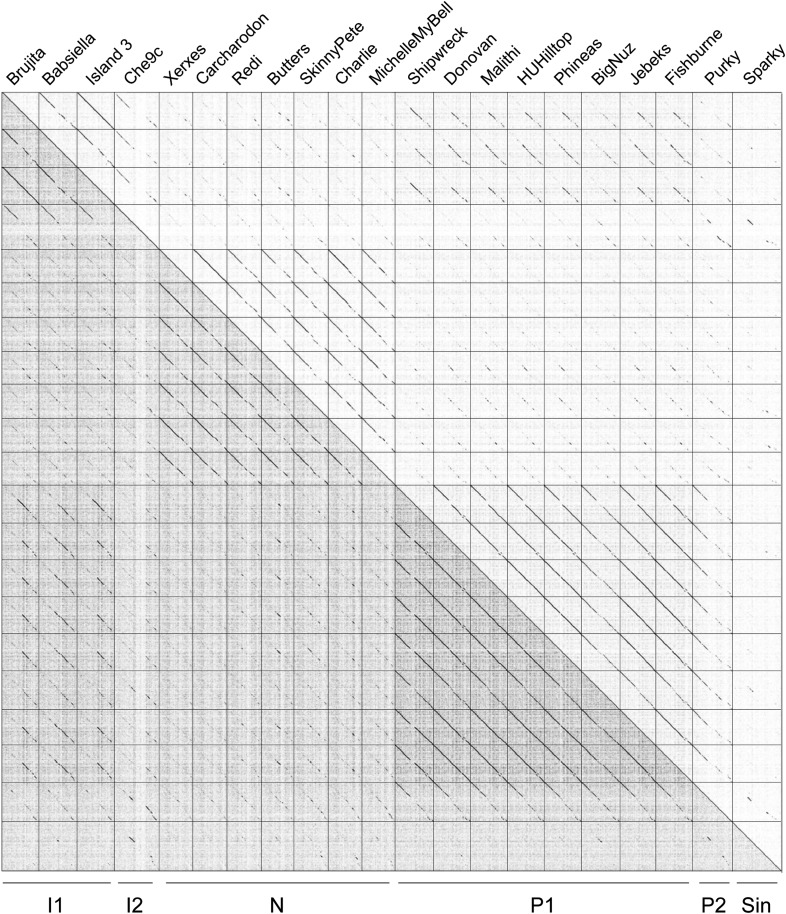
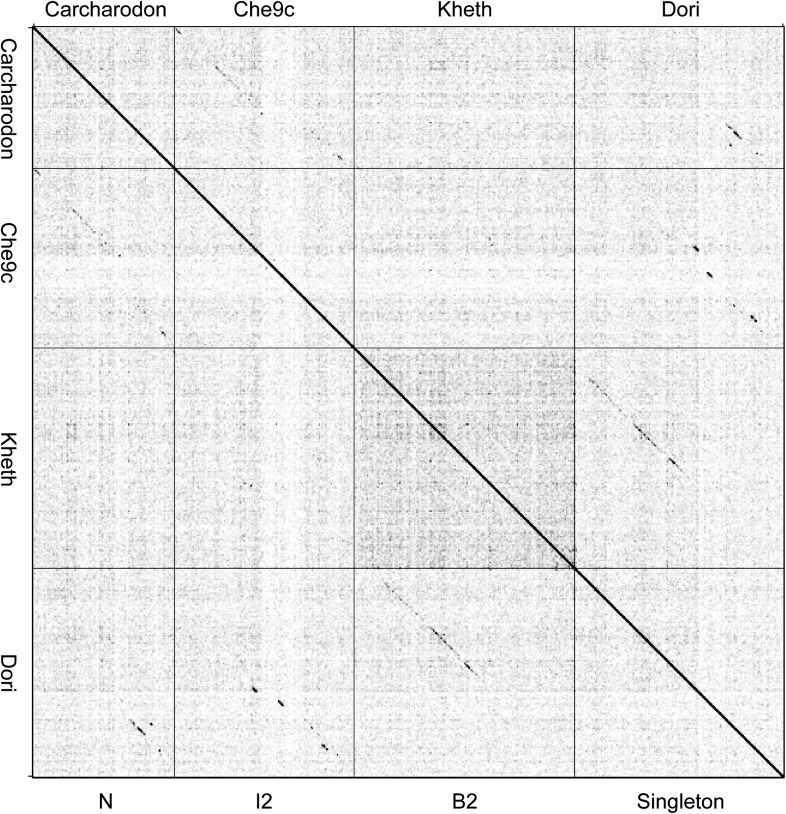
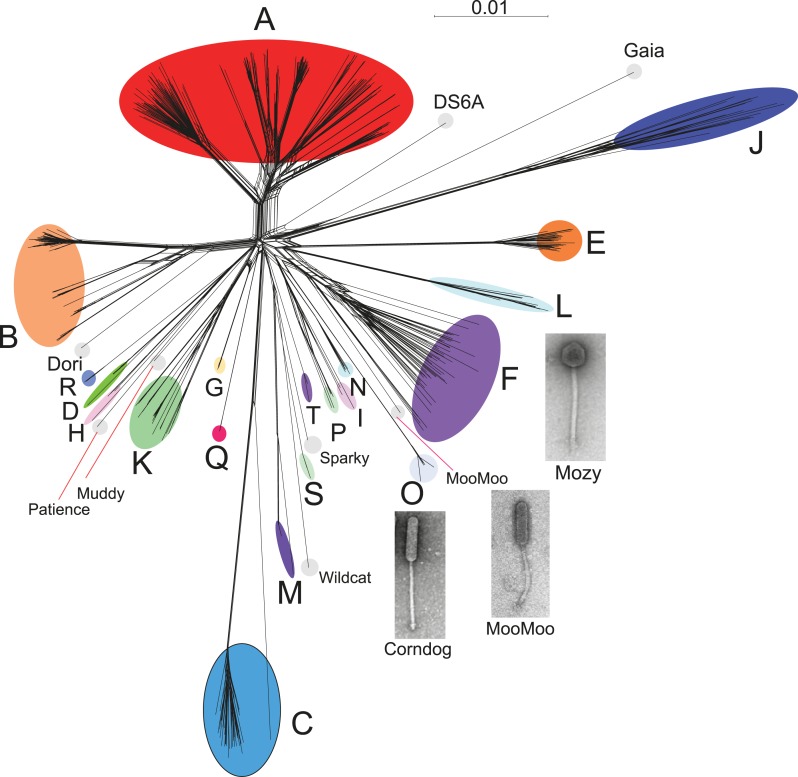
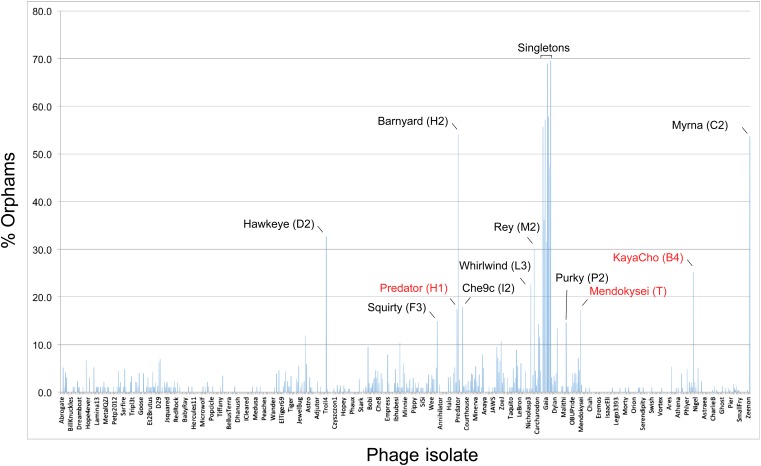
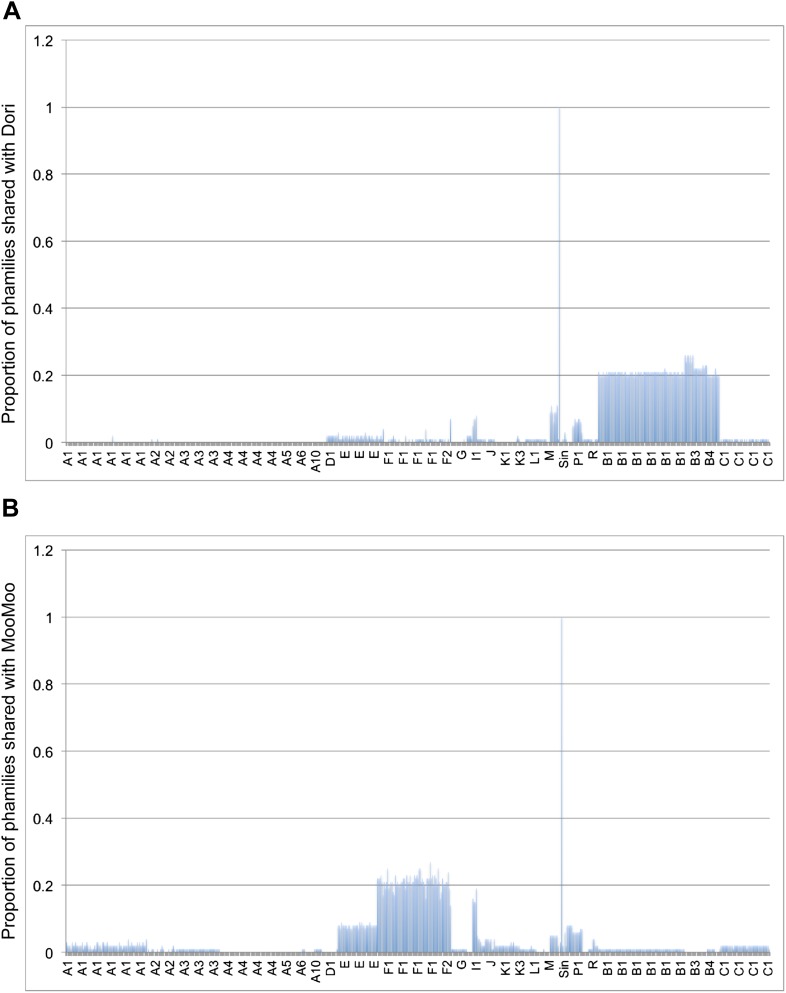
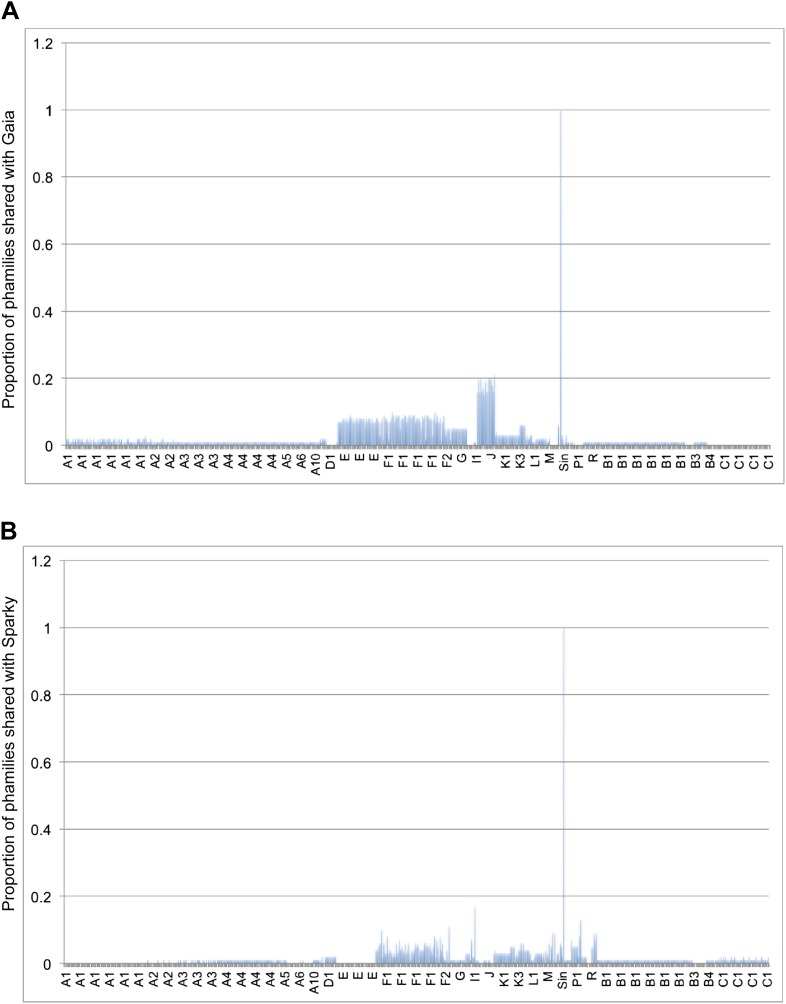
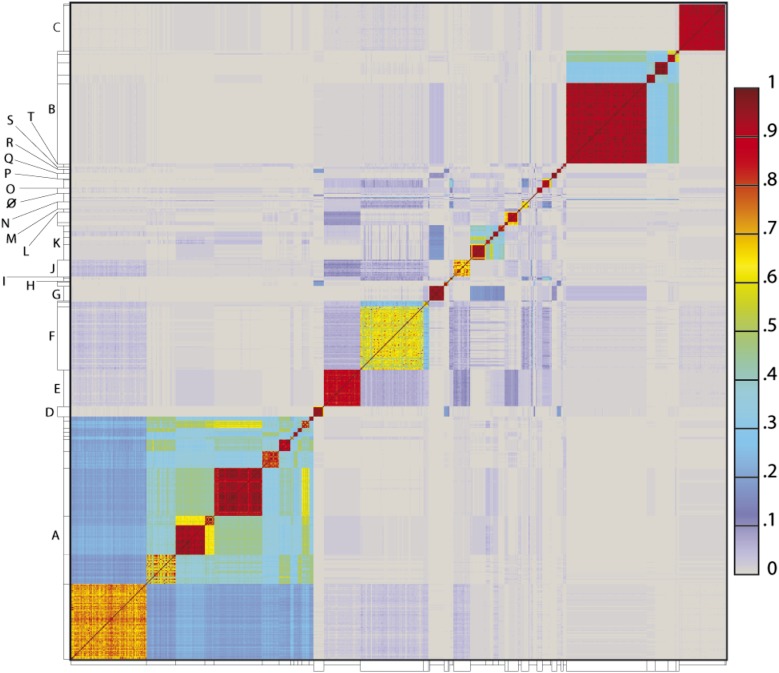
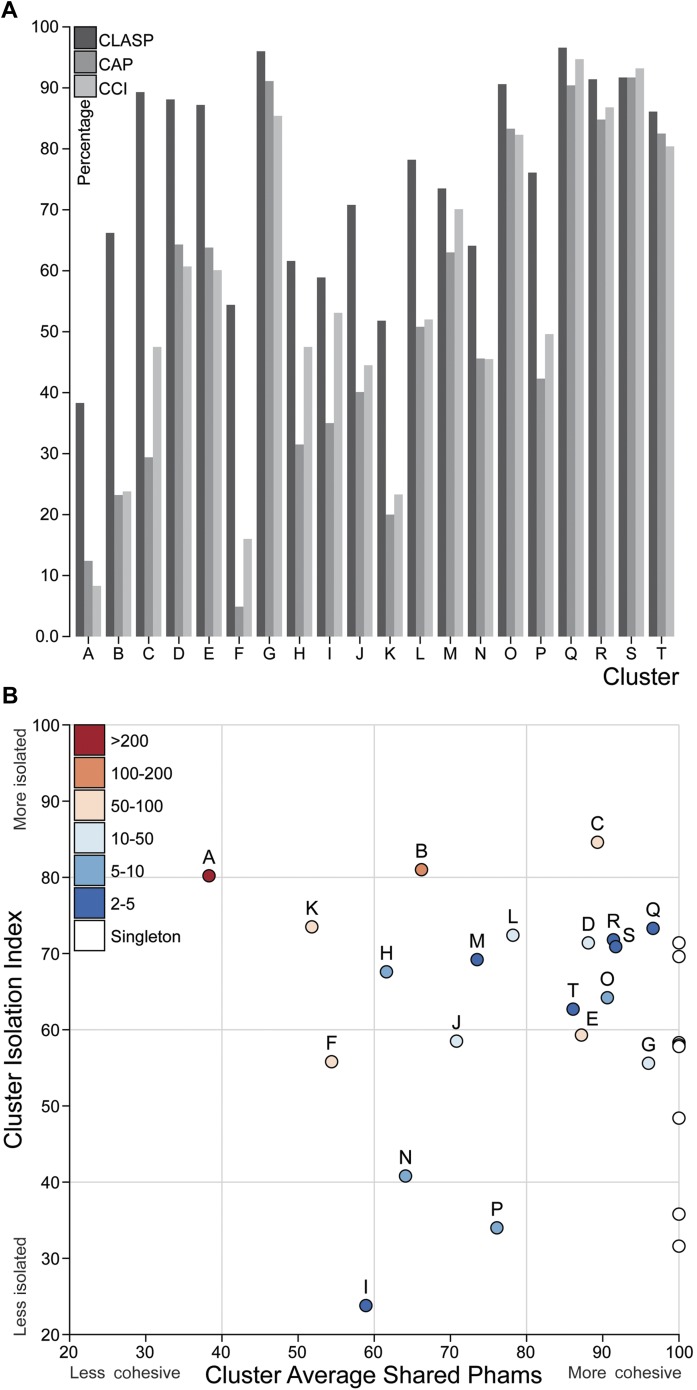
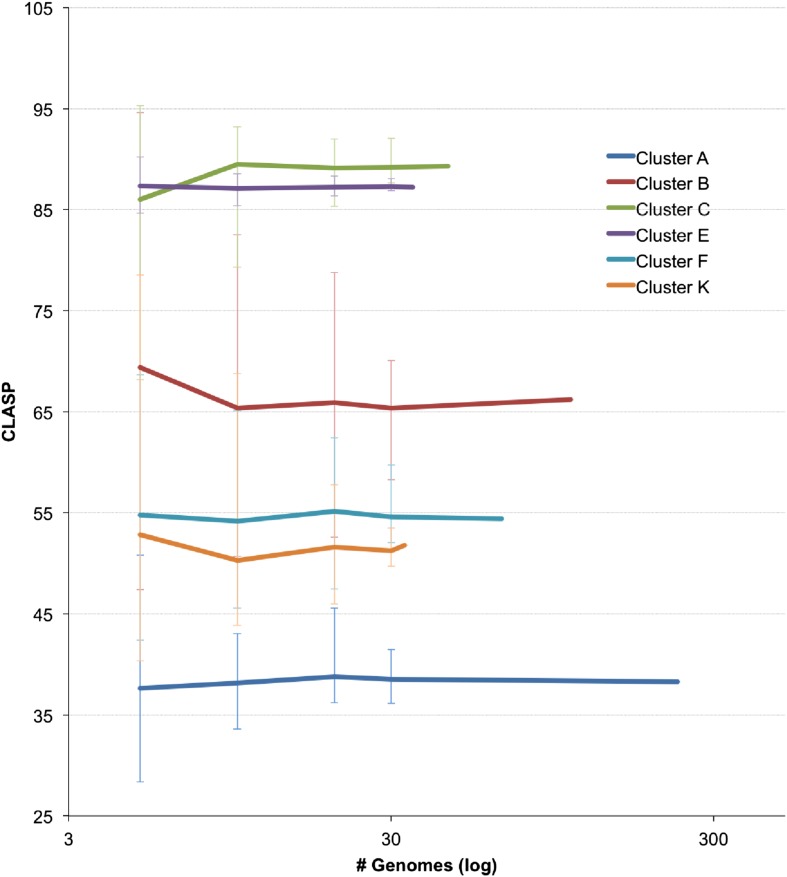
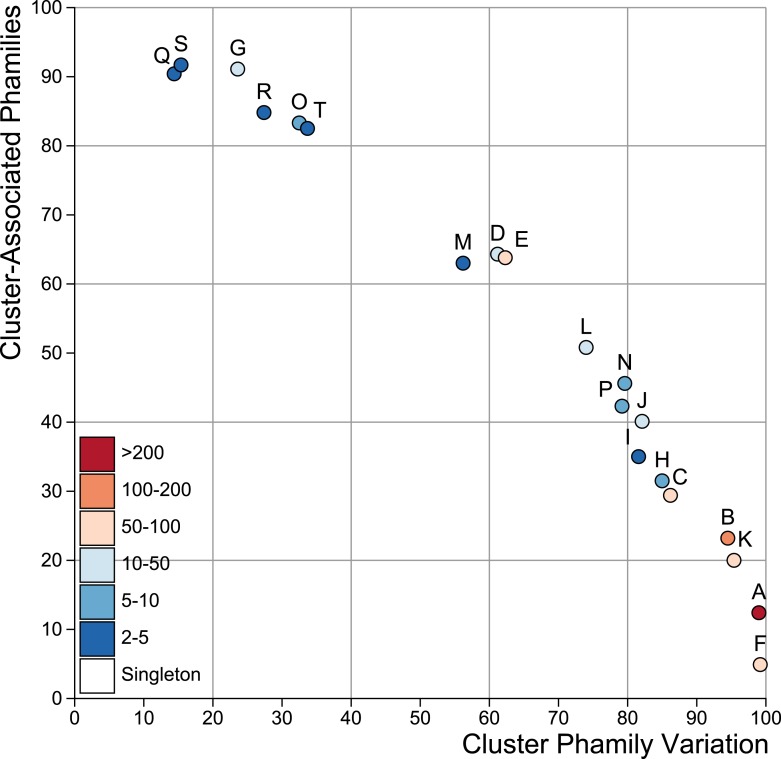
The bacteriophage population is large, dynamic, ancient, and genetically diverse. Limited genomic information shows that phage genomes are mosaic, and the genetic architecture of phage populations remains ill-defined. To understand the population structure of phages infecting a single host strain, we isolated, sequenced, and compared 627 phages of Mycobacterium smegmatis. Their genetic diversity is considerable, and there are 28 distinct genomic types (clusters) with related nucleotide sequences. However, amino acid sequence comparisons show pervasive genomic mosaicism, and quantification of inter-cluster and intra-cluster relatedness reveals a continuum of genetic diversity, albeit with uneven representation of different phages. Furthermore, rarefaction analysis shows that the mycobacteriophage population is not closed, and there is a constant influx of genes from other sources. Phage isolation and analysis was performed by a large consortium of academic institutions, illustrating the substantial benefits of a disseminated, structured program involving large numbers of freshman undergraduates in scientific discovery.
Keywords: bacteriophage; evolution; evolutionary biology; genomics; infectious disease; microbiology; viruses.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

Similar articles
-
Characterization of mycobacteria and mycobacteriophages isolated from compost at the São Paulo Zoo Park Foundation in Brazil and creation of the new mycobacteriophage Cluster U.BMC Microbiol. 2016 Jun 17;16(1):111. doi: 10.1186/s12866-016-0734-3. BMC Microbiol. 2016. PMID: 27316672 Free PMC article.
-
Bacteriophages of Gordonia spp. Display a Spectrum of Diversity and Genetic Relationships.mBio. 2017 Aug 15;8(4):e01069-17. doi: 10.1128/mBio.01069-17. mBio. 2017. PMID: 28811342 Free PMC article.
-
Cluster M mycobacteriophages Bongo, PegLeg, and Rey with unusually large repertoires of tRNA isotypes.J Virol. 2014 Mar;88(5):2461-80. doi: 10.1128/JVI.03363-13. Epub 2013 Dec 11. J Virol. 2014. PMID: 24335314 Free PMC article.
-
On the nature of mycobacteriophage diversity and host preference.Virology. 2012 Dec 20;434(2):187-201. doi: 10.1016/j.virol.2012.09.026. Epub 2012 Oct 22. Virology. 2012. PMID: 23084079 Free PMC article. Review.
-
Mycobacteriophages: genes and genomes.Annu Rev Microbiol. 2010;64:331-56. doi: 10.1146/annurev.micro.112408.134233. Annu Rev Microbiol. 2010. PMID: 20528690 Review.
Cited by
-
Construction and characterization of DNA libraries from cultured phages and environmental viromes.Appl Environ Microbiol. 2024 Oct 23;90(10):e0117124. doi: 10.1128/aem.01171-24. Epub 2024 Sep 24. Appl Environ Microbiol. 2024. PMID: 39315792 Free PMC article.
-
Complete genomes of two cluster AK Arthrobacter phages isolated from soil samples in Newburgh, NY, United States.Microbiol Resour Announc. 2024 Oct 10;13(10):e0071624. doi: 10.1128/mra.00716-24. Epub 2024 Sep 12. Microbiol Resour Announc. 2024. PMID: 39264183 Free PMC article.
-
A Bioinformatic Ecosystem for Bacteriophage Genomics: PhaMMSeqs, Phamerator, pdm_utils, PhagesDB, DEPhT, and PhamClust.Viruses. 2024 Aug 10;16(8):1278. doi: 10.3390/v16081278. Viruses. 2024. PMID: 39205252 Free PMC article. Review.
-
Comparative Genomic Analysis of Prophages in Human Vaginal Isolates of Streptococcus agalactiae.Pathogens. 2024 Jul 23;13(8):610. doi: 10.3390/pathogens13080610. Pathogens. 2024. PMID: 39204211 Free PMC article.
-
Characterization of the Structural Requirements for the NADase Activity of Bacterial Toll/IL-1R domains in a Course-based Undergraduate Research Experience.Immunohorizons. 2024 Aug 1;8(8):563-576. doi: 10.4049/immunohorizons.2300062. Immunohorizons. 2024. PMID: 39172026 Free PMC article.
References
-
- Cresawn SG, Pope WH, Jacobs-Sera D, Bowman CA, Russell DA, Dedrick RA, Adair T, Anders KR, Ball S, Bollivar D, Breitenberger C, Burnett SH, Butela K, Byrnes D, Carzo S, Cornely KA, Cross T, Daniels RL, Dunbar D, Findley AM, Gissendanner CR, Golebiewska UP, Hartzog GA, Hatherill JR, Hughes LE, Jalloh CS, De Los Santos C, Ekanam K, Khambule SL, King RA, King-Smith C, Klyczek K, Krukonis GP, Laing C, Lapin JS, Lopez AJ, Mkhwanazi SM, Molloy SD, Moran D, Munsamy V, Pacey E, Plymale R, Poxleitner M, Reyna N, Schildbach JF, Stukey J, Taylor SE, Ware VC, Wellmann AL, Westholm D, Wodarski D, Zajko M, Zikalala TS, Hendrix RW, Hatfull GF. Comparative genomics of cluster O mycobacteriophages. PLOS ONE. 2015;10:e0118725. doi: 10.1371/journal.pone.0118725. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM103430/GM/NIGMS NIH HHS/United States
- SC1 AI098976/AI/NIAID NIH HHS/United States
- GM51975/GM/NIGMS NIH HHS/United States
- GM103408/GM/NIGMS NIH HHS/United States
- R15 GM110578/GM/NIGMS NIH HHS/United States
- R01 GM051975/GM/NIGMS NIH HHS/United States
- P20 GM103430/GM/NIGMS NIH HHS/United States
- P20 GM103408/GM/NIGMS NIH HHS/United States
- GM1003423/GM/NIGMS NIH HHS/United States
- T32 GM008490/GM/NIGMS NIH HHS/United States
- 52007054/Howard Hughes Medical Institute/United States
- 54308198/Howard Hughes Medical Institute/United States
- GM094712/GM/NIGMS NIH HHS/United States
- P20 GM103423/GM/NIGMS NIH HHS/United States
- R25 GM090084/GM/NIGMS NIH HHS/United States
- R15 GM094712/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
