

Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency
- PMID: 25918518
- PMCID: PMC4394698
- DOI: 10.3389/fgene.2015.00123
Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency
Abstract
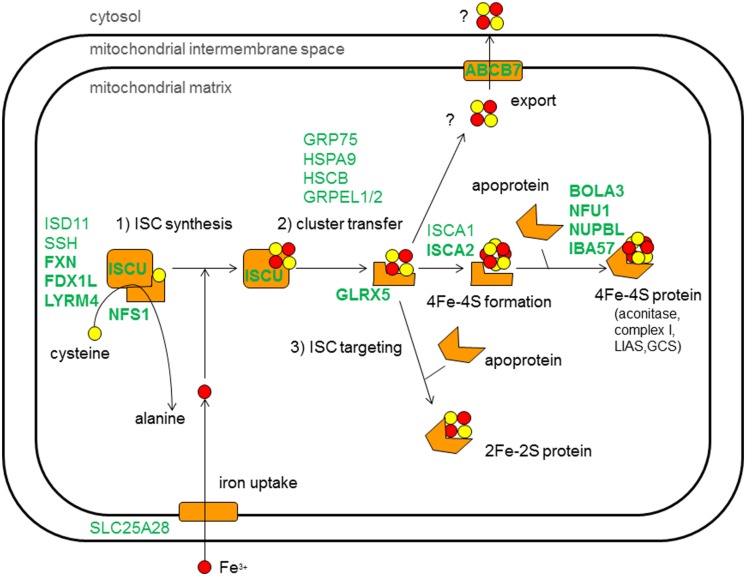
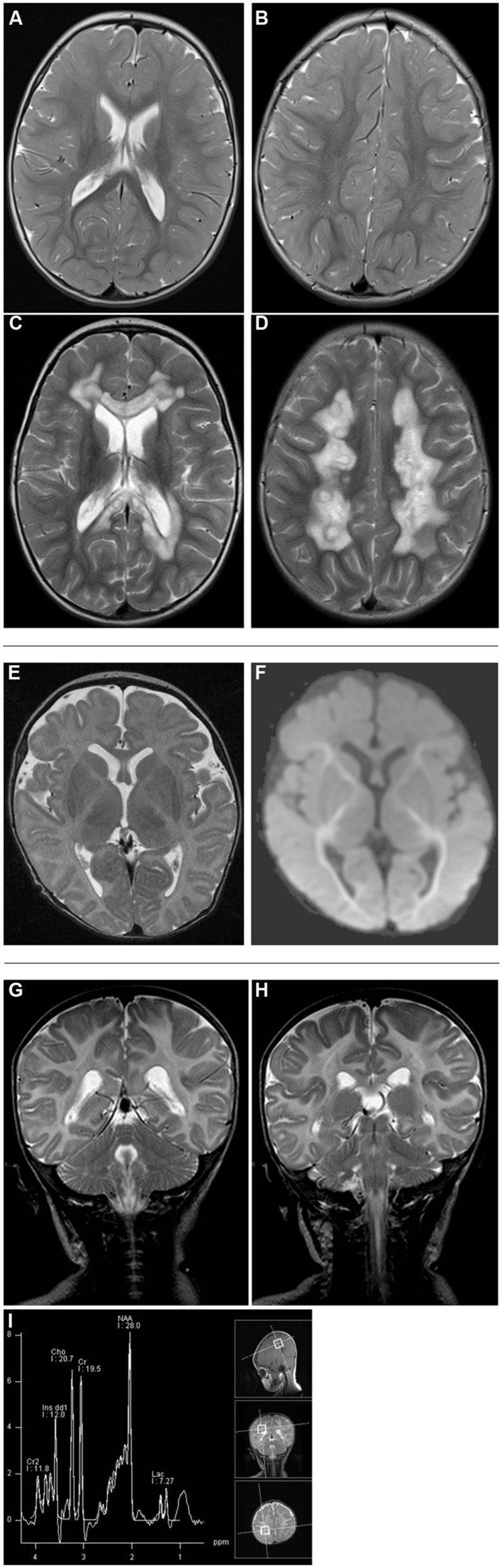
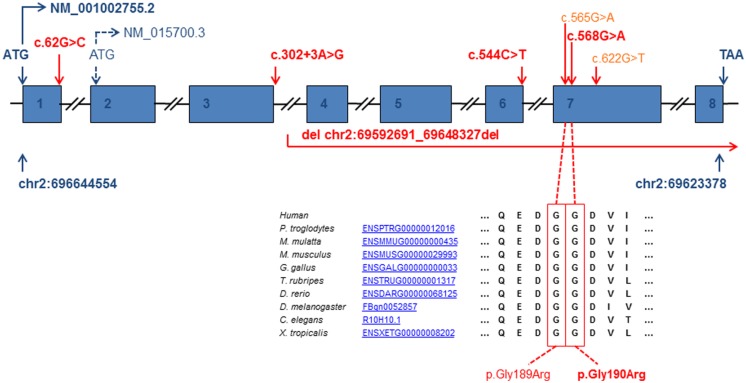
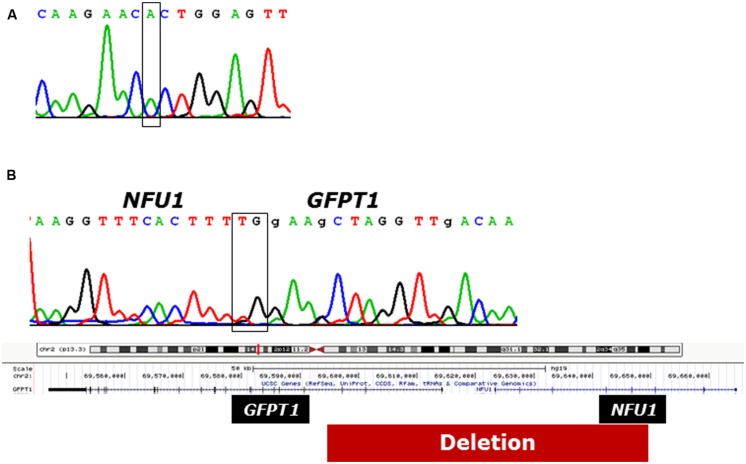
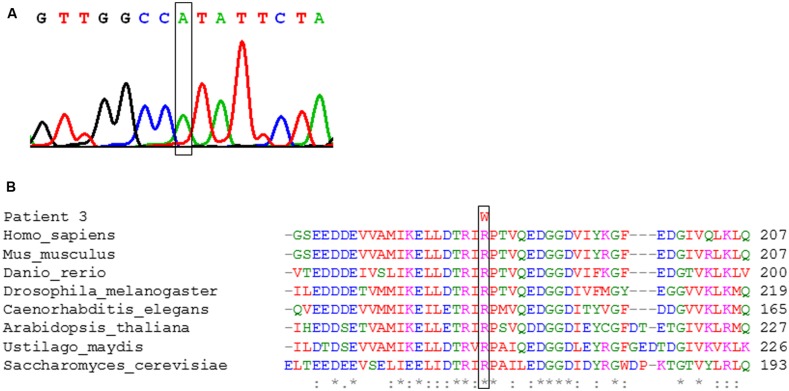
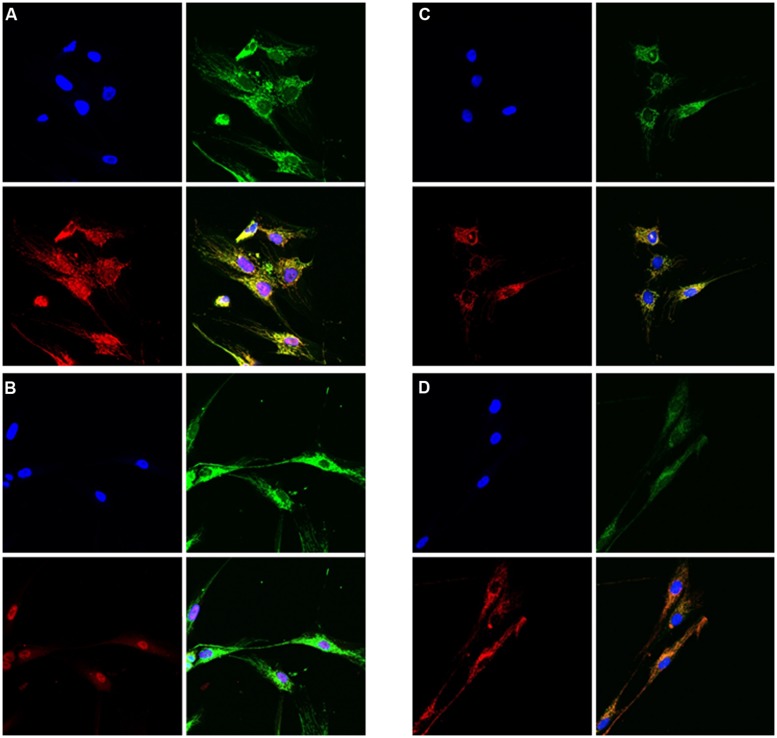
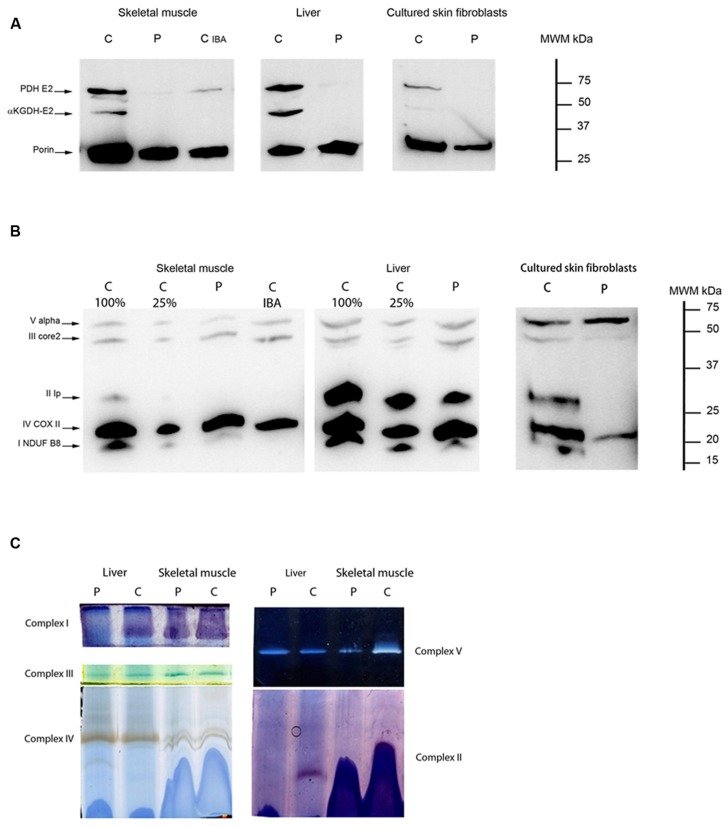
Disorders of the mitochondrial energy metabolism are clinically and genetically heterogeneous. An increasingly recognized subgroup is caused by defective mitochondrial iron-sulfur (Fe-S) cluster biosynthesis, with defects in 13 genes being linked to human disease to date. Mutations in three of them, NFU1, BOLA3, and IBA57, affect the assembly of mitochondrial [4Fe-4S] proteins leading to an impairment of diverse mitochondrial metabolic pathways and ATP production. Patients with defects in these three genes present with lactic acidosis, hyperglycinemia, and reduced activities of respiratory chain complexes I and II, the four lipoic acid-dependent 2-oxoacid dehydrogenases and the glycine cleavage system (GCS). To date, five different NFU1 pathogenic variants have been reported in 15 patients from 12 families. We report on seven new patients from five families carrying compound heterozygous or homozygous pathogenic NFU1 mutations identified by candidate gene screening and exome sequencing. Six out of eight different disease alleles were novel and functional studies were performed to support the pathogenicity of five of them. Characteristic clinical features included fatal infantile encephalopathy and pulmonary hypertension leading to death within the first 6 months of life in six out of seven patients. Laboratory investigations revealed combined defects of pyruvate dehydrogenase complex (five out of five) and respiratory chain complexes I and II+III (four out of five) in skeletal muscle and/or cultured skin fibroblasts as well as increased lactate (five out of six) and glycine concentration (seven out of seven). Our study contributes to a better definition of the phenotypic spectrum associated with NFU1 mutations and to the diagnostic workup of future patients.
Keywords: NFU1; iron–sulfur cluster; lipoic acid; mitochondrial respiratory chain; pulmonary hypertension.
Figures
Similar articles
-
A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation.Biomedicines. 2021 Aug 10;9(8):989. doi: 10.3390/biomedicines9080989. Biomedicines. 2021. PMID: 34440194 Free PMC article. Review.
-
Impact of mutations within the [Fe-S] cluster or the lipoic acid biosynthesis pathways on mitochondrial protein expression profiles in fibroblasts from patients.Mol Genet Metab. 2017 Nov;122(3):85-94. doi: 10.1016/j.ymgme.2017.08.001. Epub 2017 Aug 3. Mol Genet Metab. 2017. PMID: 28803783
-
A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins.Am J Hum Genet. 2011 Nov 11;89(5):656-67. doi: 10.1016/j.ajhg.2011.10.005. Am J Hum Genet. 2011. PMID: 22077971 Free PMC article.
-
Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes.Hum Mol Genet. 2014 Apr 1;23(7):1907-15. doi: 10.1093/hmg/ddt585. Epub 2013 Nov 20. Hum Mol Genet. 2014. PMID: 24256811
-
Lipoic acid biosynthesis defects.J Inherit Metab Dis. 2014 Jul;37(4):553-63. doi: 10.1007/s10545-014-9705-8. Epub 2014 Apr 29. J Inherit Metab Dis. 2014. PMID: 24777537 Review.
Cited by
-
Loss-of-function mutations in ISCA2 disrupt 4Fe-4S cluster machinery and cause a fatal leukodystrophy with hyperglycinemia and mtDNA depletion.Hum Mutat. 2018 Apr;39(4):537-549. doi: 10.1002/humu.23396. Epub 2018 Jan 22. Hum Mutat. 2018. PMID: 29297947 Free PMC article.
-
Hypoxia and the integrated stress response promote pulmonary hypertension and preeclampsia: Implications in drug development.Drug Discov Today. 2021 Nov;26(11):2754-2773. doi: 10.1016/j.drudis.2021.07.011. Epub 2021 Jul 22. Drug Discov Today. 2021. PMID: 34302972 Free PMC article. Review.
-
Clinical Implications of the Genetic Background in Pediatric Pulmonary Arterial Hypertension: Data from the Spanish REHIPED Registry.Int J Mol Sci. 2022 Sep 9;23(18):10433. doi: 10.3390/ijms231810433. Int J Mol Sci. 2022. PMID: 36142358 Free PMC article.
-
A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation.Biomedicines. 2021 Aug 10;9(8):989. doi: 10.3390/biomedicines9080989. Biomedicines. 2021. PMID: 34440194 Free PMC article. Review.
-
Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension.Antioxidants (Basel). 2022 Feb 21;11(2):428. doi: 10.3390/antiox11020428. Antioxidants (Basel). 2022. PMID: 35204311 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous