

Channelopathies of skeletal muscle excitability
- PMID: 25880512
- PMCID: PMC4754081
- DOI: 10.1002/cphy.c140062
Channelopathies of skeletal muscle excitability
Abstract
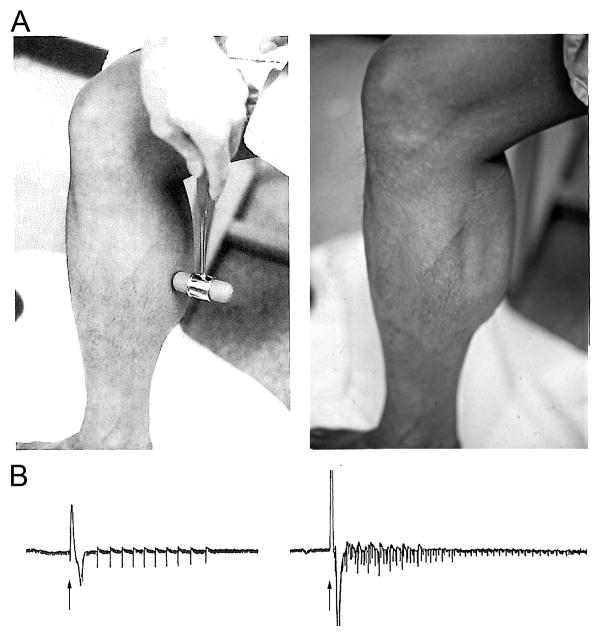
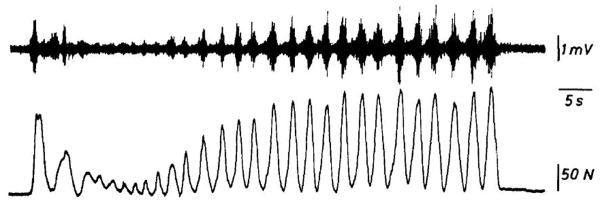
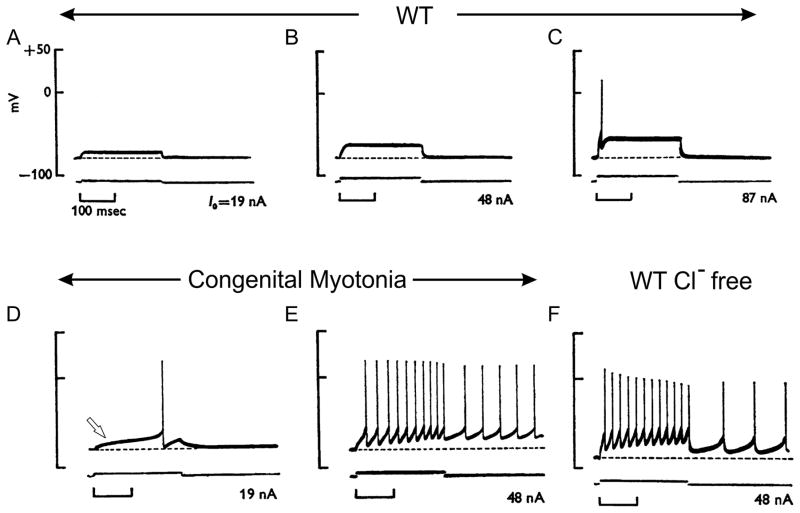
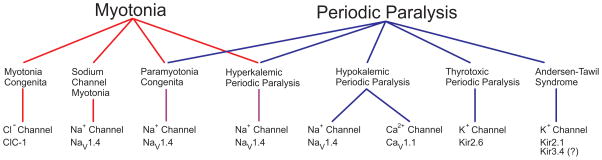
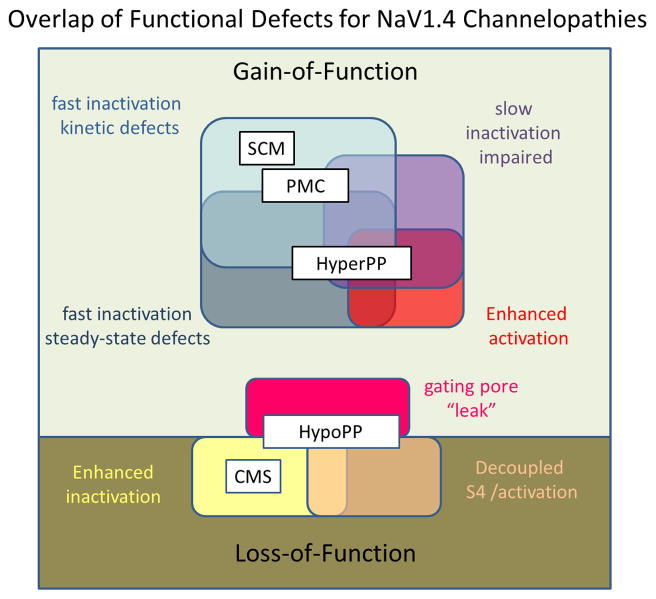
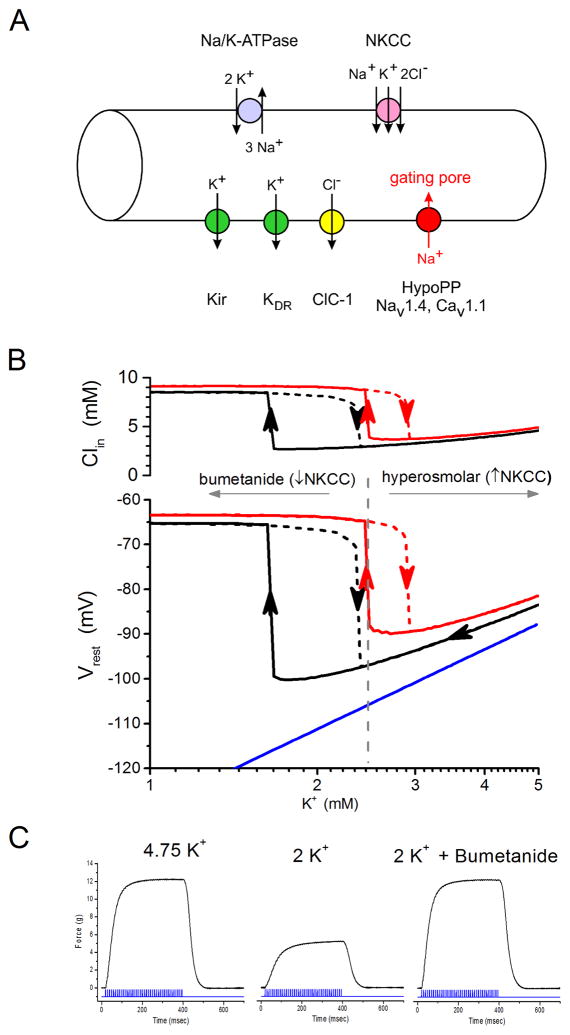
Familial disorders of skeletal muscle excitability were initially described early in the last century and are now known to be caused by mutations of voltage-gated ion channels. The clinical manifestations are often striking, with an inability to relax after voluntary contraction (myotonia) or transient attacks of severe weakness (periodic paralysis). An essential feature of these disorders is fluctuation of symptoms that are strongly impacted by environmental triggers such as exercise, temperature, or serum K(+) levels. These phenomena have intrigued physiologists for decades, and in the past 25 years the molecular lesions underlying these disorders have been identified and mechanistic studies are providing insights for therapeutic strategies of disease modification. These familial disorders of muscle fiber excitability are "channelopathies" caused by mutations of a chloride channel (ClC-1), sodium channel (NaV1.4), calcium channel (CaV1.1), and several potassium channels (Kir2.1, Kir2.6, and Kir3.4). This review provides a synthesis of the mechanistic connections between functional defects of mutant ion channels, their impact on muscle excitability, how these changes cause clinical phenotypes, and approaches toward therapeutics.
© 2015 American Physiological Society.
Figures

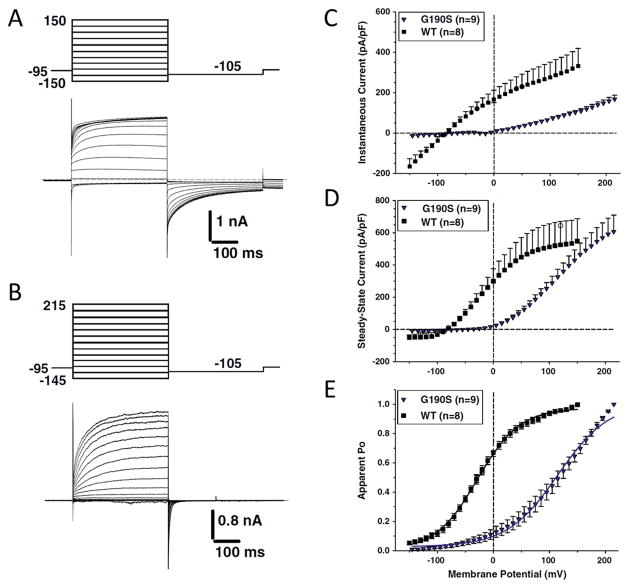
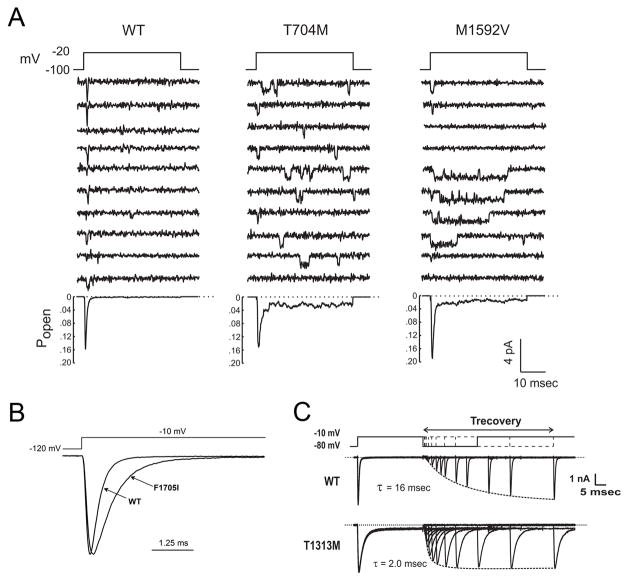
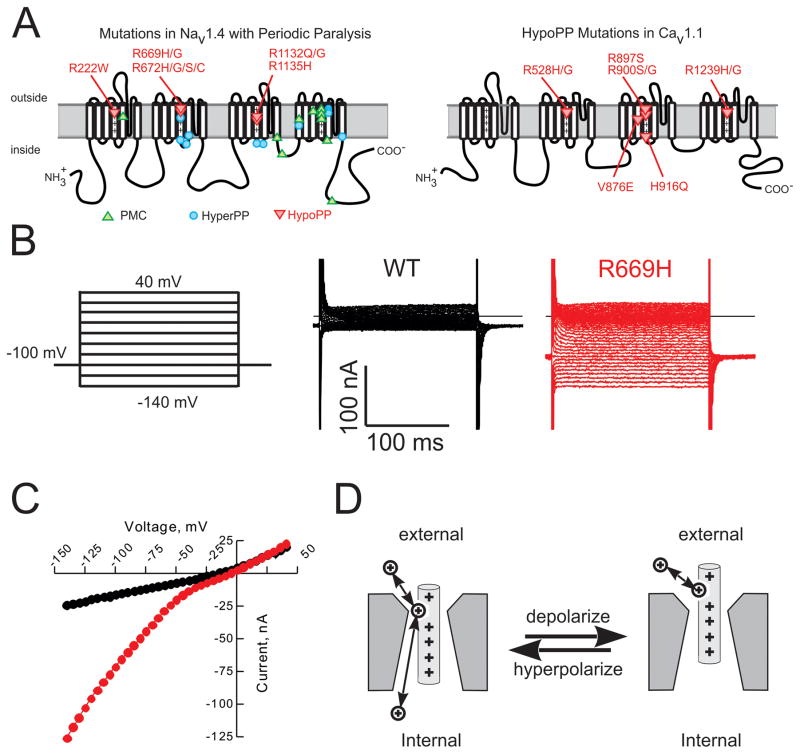
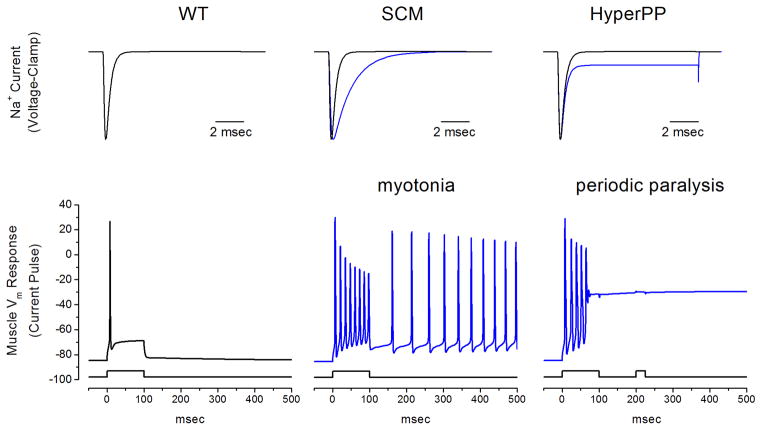
Similar articles
-
Voltage-sensor mutations in channelopathies of skeletal muscle.J Physiol. 2010 Jun 1;588(Pt 11):1887-95. doi: 10.1113/jphysiol.2010.186874. Epub 2010 Feb 15. J Physiol. 2010. PMID: 20156847 Free PMC article. Review.
-
Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia.Neurology. 2008 Mar 4;70(10):755-61. doi: 10.1212/01.wnl.0000265397.70057.d8. Epub 2007 Sep 26. Neurology. 2008. PMID: 17898326 Free PMC article.
-
Periodic paralysis.Handb Clin Neurol. 2018;148:505-520. doi: 10.1016/B978-0-444-64076-5.00032-6. Handb Clin Neurol. 2018. PMID: 29478596 Review.
-
Pathomechanisms in channelopathies of skeletal muscle and brain.Annu Rev Neurosci. 2006;29:387-415. doi: 10.1146/annurev.neuro.29.051605.112815. Annu Rev Neurosci. 2006. PMID: 16776591 Review.
-
Muscle biopsy and cell cultures: potential diagnostic tools in hereditary skeletal muscle channelopathies.Eur J Histochem. 2003;47(1):17-28. doi: 10.4081/803. Eur J Histochem. 2003. PMID: 12685554
Cited by
-
Mice with an NaV1.4 sodium channel null allele have latent myasthenia, without susceptibility to periodic paralysis.Brain. 2016 Jun;139(Pt 6):1688-99. doi: 10.1093/brain/aww070. Epub 2016 Apr 5. Brain. 2016. PMID: 27048647 Free PMC article.
-
Unveiling the Multifaceted Problems Associated with Dysrhythmia.Int J Mol Sci. 2023 Dec 23;25(1):263. doi: 10.3390/ijms25010263. Int J Mol Sci. 2023. PMID: 38203440 Free PMC article. Review.
-
Skeletal Muscle Channelopathies.Neurotherapeutics. 2018 Oct;15(4):954-965. doi: 10.1007/s13311-018-00678-0. Neurotherapeutics. 2018. PMID: 30341599 Free PMC article. Review.
-
Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation.Sci Rep. 2019 Nov 26;9(1):17560. doi: 10.1038/s41598-019-54041-0. Sci Rep. 2019. PMID: 31772215 Free PMC article.
-
Substitutions of the S4DIV R2 residue (R1451) in NaV1.4 lead to complex forms of paramyotonia congenita and periodic paralyses.Sci Rep. 2018 Feb 1;8(1):2041. doi: 10.1038/s41598-018-20468-0. Sci Rep. 2018. PMID: 29391559 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
