

A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques
- PMID: 25853458
- PMCID: PMC4382926
- DOI: 10.1186/s40478-015-0190-6
A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques
Abstract
Introduction: Although TDP-43 is the main constituent of the ubiquitinated cytoplasmic inclusions in the most common forms of frontotemporal lobar degeneration, TARDBP mutations are not a common cause of familial frontotemporal dementia, especially in the absence of motor neuron disease.
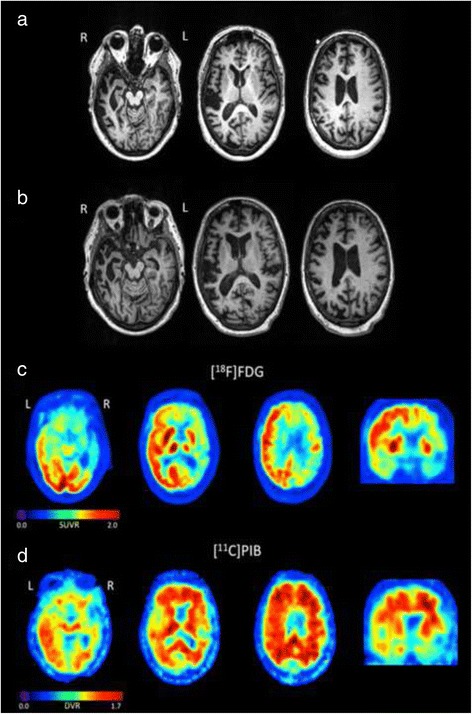
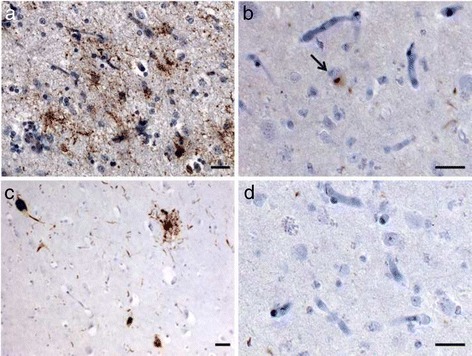
Results: We describe a pedigree presenting with a complex autosomal dominant disease, with a heterogeneous clinical phenotype, comprising unspecified dementia, parkinsonism, frontotemporal dementia and motor neuron disease. Genetic analyses identified a novel P112H TARDBP double variation located in exon 3 coding for the first RNA recognition motif of the protein (RRM1). This double mutation is probably pathogenic based on neuropathological findings, in silico prediction analysis and exome sequencing. The two autopsied siblings described here presented with frontotemporal dementia involving multiple cognitive domains and behavior but lacking symptoms of motor neuron disease throughout the disease course. The siblings presented with strikingly similar, although atypical, neuropathological features, including an unclassifiable TDP-43 inclusion pattern, a high burden of tau-negative β-amyloid neuritic plaques with an AD-like biochemical profile, and an unclassifiable 4-repeat tauopathy. The co-occurrence of multiple protein inclusions points to a pathogenic mechanism that facilitates misfolded protein interaction and aggregation or a loss of TDP-43 function that somehow impairs protein clearance.
Conclusions: TARDBP mutation screening should be considered in familial frontotemporal dementia cases, even without signs or symptoms of motor neuron disease, especially when other more frequent causes of genetic frontotemporal dementia (i.e. GRN, C9ORF72, MAPT) have been excluded and when family history is complex and includes parkinsonism, motor neuron disease and frontotemporal dementia. Further investigations in this family may provide insight into the physiological functions of TARDBP.
Figures




Similar articles
-
Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features.Brain. 2012 Mar;135(Pt 3):736-50. doi: 10.1093/brain/awr361. Brain. 2012. PMID: 22366791 Free PMC article.
-
Heterogeneous ribonuclear protein A3 (hnRNP A3) is present in dipeptide repeat protein containing inclusions in Frontotemporal Lobar Degeneration and Motor Neurone disease associated with expansions in C9orf72 gene.Acta Neuropathol Commun. 2017 Apr 21;5(1):31. doi: 10.1186/s40478-017-0437-5. Acta Neuropathol Commun. 2017. PMID: 28431575 Free PMC article.
-
Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease.Ann Neurol. 2010 Nov;68(5):639-49. doi: 10.1002/ana.22274. Ann Neurol. 2010. PMID: 21031579
-
Frontotemporal lobar degeneration. An update on clinical, pathological and genetic findings.Gerontology. 2001 Jan-Feb;47(1):1-8. doi: 10.1159/000052763. Gerontology. 2001. PMID: 11244285 Review.
-
A novel frameshift GRN mutation results in frontotemporal lobar degeneration with a distinct clinical phenotype in two siblings: case report and literature review.BMC Neurol. 2017 Sep 15;17(1):182. doi: 10.1186/s12883-017-0959-2. BMC Neurol. 2017. PMID: 28915852 Free PMC article. Review.
Cited by
-
Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis.Front Mol Neurosci. 2019 Feb 14;12:25. doi: 10.3389/fnmol.2019.00025. eCollection 2019. Front Mol Neurosci. 2019. PMID: 30837838 Free PMC article. Review.
-
Cytoplasmic Relocalization of TAR DNA-Binding Protein 43 Is Not Sufficient to Reproduce Cellular Pathologies Associated with ALS In vitro.Front Mol Neurosci. 2017 Feb 24;10:46. doi: 10.3389/fnmol.2017.00046. eCollection 2017. Front Mol Neurosci. 2017. PMID: 28286471 Free PMC article.
-
Right temporal variant frontotemporal dementia is pathologically heterogeneous: a case-series and a systematic review.Acta Neuropathol Commun. 2021 Aug 3;9(1):131. doi: 10.1186/s40478-021-01229-z. Acta Neuropathol Commun. 2021. PMID: 34344452 Free PMC article.
-
Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies.Pharmaceuticals (Basel). 2024 Oct 18;17(10):1391. doi: 10.3390/ph17101391. Pharmaceuticals (Basel). 2024. PMID: 39459030 Free PMC article. Review.
-
Aggregation of the nucleic acid-binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation.J Biol Chem. 2019 Mar 8;294(10):3696-3706. doi: 10.1074/jbc.RA118.006351. Epub 2019 Jan 10. J Biol Chem. 2019. PMID: 30630951 Free PMC article.
References
-
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Muñoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, Consortium for Frontotemporal Lobar Degeneration Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. - DOI - PMC - PubMed
-
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
