

Whole Exome Sequencing Reveals DYSF, FKTN, and ISPD Mutations in Congenital Muscular Dystrophy Without Brain or Eye Involvement
- PMID: 25821721
- PMCID: PMC4373448
- DOI: 10.3233/JND-140038
Whole Exome Sequencing Reveals DYSF, FKTN, and ISPD Mutations in Congenital Muscular Dystrophy Without Brain or Eye Involvement
Abstract
Background: Congenital muscular dystrophies (CMDs) are a genetically and clinically heterogeneous group of neuromuscular disorders. Several genes encoding extracellular matrix, nuclear envelope, sarcolemmal proteins and glycosylation enzymes have been implicated in CMDs. The large overlap of clinical presentations due to mutations in different genes poses a challenge for clinicians in determining disease etiology for each patient.
Objective: We investigated the use of whole exome sequencing (WES) in identifying the genetic cause of disease in 5 CMD patients from 3 families who presented with highly similar clinical features, including early-onset rapidly progressive weakness without brain or eye abnormalities.
Methods: Whole exome sequencing was performed on DNA from affected individuals. Potential functional impacts of mutations were investigated by immunostaining on available muscle biopsies.
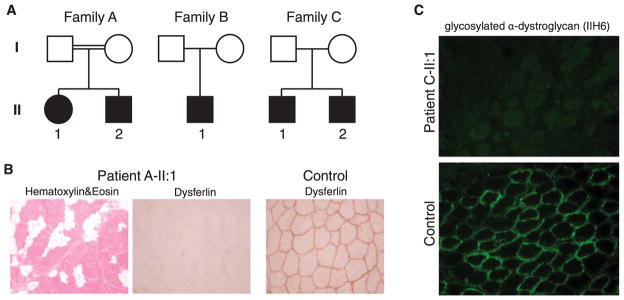
Results: Pathogenic mutations in 3 different genes, DYSF, FKTN, and ISPD were identified in each family. Mutation in DYSF led to absence of dysferlin protein in patient muscle. Mutations in ISPD led to impaired ISDP function, as demonstrated by deficiency of α-dystroglycan glycosylation in patient muscle.
Conclusions: This study highlights the benefit of unbiased genomic approaches in molecular diagnosis of neuromuscular disorders with high clinical heterogeneity, such as the phenotypes observed in our patients. Our results suggest that dysferlin deficiency should be in the differential diagnosis of congenital and rapidly progressive muscular dystrophy, and therefore dysferlin antibody should be in the standard immunohistochemistry panel for muscle biopsies in cases with suspected CMD.
Keywords: DYSF protein human; Dysferlinopathy; FKTN protein human; ISPD protein human; Muscular dystrophies; genetic testing; neuromuscular diseases.
Conflict of interest statement
The authors have no conflicts of interest to report.
Figures
Similar articles
-
Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned.JAMA Neurol. 2015 Dec;72(12):1424-32. doi: 10.1001/jamaneurol.2015.2274. JAMA Neurol. 2015. PMID: 26436962
-
Genetic profile for suspected dysferlinopathy identified by targeted next-generation sequencing.Neurol Genet. 2015 Dec 10;1(4):e36. doi: 10.1212/NXG.0000000000000036. eCollection 2015 Dec. Neurol Genet. 2015. PMID: 27066573 Free PMC article.
-
Whole-exome analyses of congenital muscular dystrophy and congenital myopathy patients from India reveal a wide spectrum of known and novel mutations.Eur J Neurol. 2021 Mar;28(3):992-1003. doi: 10.1111/ene.14616. Epub 2020 Nov 26. Eur J Neurol. 2021. PMID: 33124102
-
[Fukuyama congenital muscular dystrophy and related alpha-dystroglycanopathies].Brain Nerve. 2008 Oct;60(10):1159-64. Brain Nerve. 2008. PMID: 18975603 Review. Japanese.
-
Defective glycosylation in muscular dystrophy.Lancet. 2002 Nov 2;360(9343):1419-21. doi: 10.1016/S0140-6736(02)11397-3. Lancet. 2002. PMID: 12424008 Review.
Cited by
-
Novel fukutin mutations in limb-girdle muscular dystrophy type 2M with childhood onset.Neurol Genet. 2017 Jul 10;3(4):e167. doi: 10.1212/NXG.0000000000000167. eCollection 2017 Aug. Neurol Genet. 2017. PMID: 28785732 Free PMC article. No abstract available.
-
Neurology--the next 10 years.Nat Rev Neurol. 2015 Nov;11(11):658-64. doi: 10.1038/nrneurol.2015.196. Epub 2015 Oct 27. Nat Rev Neurol. 2015. PMID: 26503922 Review.
-
Genetic cause of heterogeneous inherited myopathies in a cohort of Greek patients.Mol Genet Metab Rep. 2020 Nov 30;25:100682. doi: 10.1016/j.ymgmr.2020.100682. eCollection 2020 Dec. Mol Genet Metab Rep. 2020. PMID: 33304817 Free PMC article.
-
Clinical Reasoning: A 30-year-old man with progressive weakness and atrophy.Neurology. 2016 Nov 8;87(19):e227-e230. doi: 10.1212/WNL.0000000000003320. Neurology. 2016. PMID: 27821570 Free PMC article. No abstract available.
References
-
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. - PubMed
-
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2–3):377–394. - PubMed
-
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–323. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
