

Glucocerebrosidase and Parkinson disease: Recent advances
- PMID: 25802027
- PMCID: PMC4471139
- DOI: 10.1016/j.mcn.2015.03.013
Glucocerebrosidase and Parkinson disease: Recent advances
Abstract
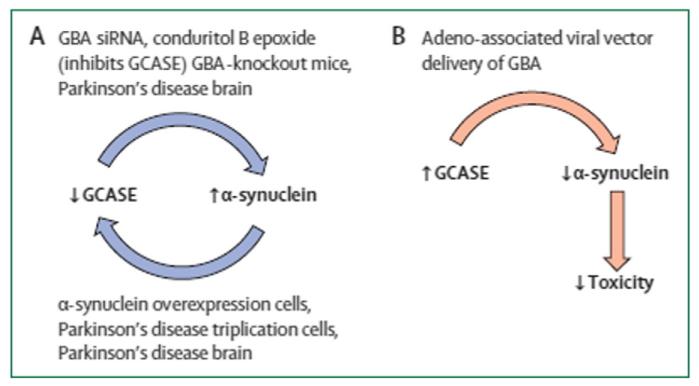
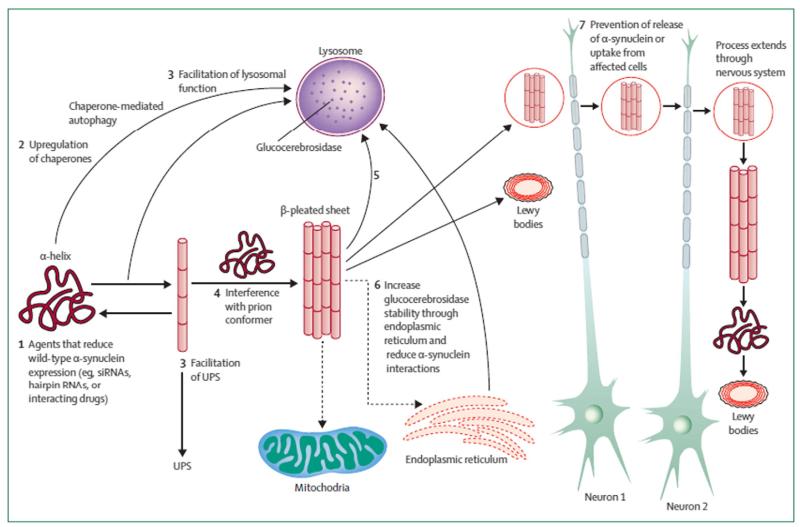
Mutations of the glucocerebrosidase (GBA) gene are the most important risk factor yet discovered for Parkinson disease (PD). Homozygous GBA mutations result in Gaucher disease (GD), a lysosomal storage disorder. Heterozygous mutations have not until recently been thought to be associated with any pathological process. However, it is clear that the presence of a GBA mutation in homozygous or heterozygous form is associated with an approximately 20-fold increase in the risk for PD, with little if any difference in risk burden related to gene dose. Most studies suggest that 5-10% of PD patients have GBA mutations, although this figure is greater in the Ashkenazi population and may be an underestimate overall if the entire exome is not sequenced. GBA-associated PD is clinically indistinguishable from idiopathic PD, except for slightly earlier age of onset and a greater frequency of cognitive impairment. Pathological and imaging features, and response to pharmacotherapy are identical to idiopathic PD. GBA mutations result in reduced enzyme activity and mutant protein may become trapped in the endoplasmic reticulum (ER) leading to unfolded protein response and ER associated degradation and stress. Both mechanisms may be relevant in GD and PD pathogenesis and lead to impaired lysosomal function. Of particular relevance to PD is the interaction of glucocerebrosidase enzyme (GCase) with alpha-synuclein (SNCA). There appears to be a bi-directional reciprocal relationship between GCase levels and those of SNCA. Thus reduced GCase in GBA mutation PD brain is associated with increased SNCA, and increased SNCA deposition is associated with reduced GCase even in GBA wild-type PD brains. It is noteworthy that GBA mutations are also associated with an increase in risk for dementia with Lewy bodies, another synucleinopathy. It has been suggested that the relationship between GCase and SNCA may be leveraged to reduce SNCA levels in PD by enhancing GCase levels and activity. This hypothesis has been confirmed in GBA mutant mice, PD patient fibroblasts and cells with SNCA overexpression, and offers an important target pathway for future neuroprotection therapy in PD. This article is part of a Special Issue entitled 'Neuronal Protein'.
Keywords: Alpha-synuclein; Glucocerebrosidase; Lysosome; Neuroprotection; Parkinson disease.
Copyright © 2015. Published by Elsevier Inc.
Figures
Similar articles
-
The relationship between glucocerebrosidase mutations and Parkinson disease.J Neurochem. 2016 Oct;139 Suppl 1(Suppl Suppl 1):77-90. doi: 10.1111/jnc.13385. Epub 2016 Feb 10. J Neurochem. 2016. PMID: 26860875 Free PMC article. Review.
-
GBA Variants and Parkinson Disease: Mechanisms and Treatments.Cells. 2022 Apr 8;11(8):1261. doi: 10.3390/cells11081261. Cells. 2022. PMID: 35455941 Free PMC article. Review.
-
Glucocerebrosidase and Parkinson Disease: Molecular, Clinical, and Therapeutic Implications.Neuroscientist. 2018 Oct;24(5):540-559. doi: 10.1177/1073858417748875. Epub 2018 Feb 4. Neuroscientist. 2018. PMID: 29400127 Review.
-
Glucocerebrosidase mutations and the pathogenesis of Parkinson disease.Ann Med. 2013 Dec;45(8):511-21. doi: 10.3109/07853890.2013.849003. Ann Med. 2013. PMID: 24219755 Review.
-
Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations.Autophagy. 2019 Jan;15(1):113-130. doi: 10.1080/15548627.2018.1509818. Epub 2018 Oct 12. Autophagy. 2019. PMID: 30160596 Free PMC article.
Cited by
-
Complicity of α-synuclein oligomer and calcium dyshomeostasis in selective neuronal vulnerability in Lewy body disease.Arch Pharm Res. 2021 Jun;44(6):564-573. doi: 10.1007/s12272-021-01334-6. Epub 2021 Jun 10. Arch Pharm Res. 2021. PMID: 34114191 Free PMC article. Review.
-
Expanding role of molecular chaperones in regulating α-synuclein misfolding; implications in Parkinson's disease.Cell Mol Life Sci. 2017 Feb;74(4):617-629. doi: 10.1007/s00018-016-2340-9. Epub 2016 Aug 13. Cell Mol Life Sci. 2017. PMID: 27522545 Free PMC article. Review.
-
The prodromes of Parkinson's disease.Eur J Neurosci. 2019 Feb;49(3):320-327. doi: 10.1111/ejn.14269. Epub 2018 Dec 5. Eur J Neurosci. 2019. PMID: 30447019 Free PMC article. Review.
-
Dermal Phospho-Alpha-Synuclein Deposition in Patients With Parkinson's Disease and Mutation of the Glucocerebrosidase Gene.Front Neurol. 2018 Dec 17;9:1094. doi: 10.3389/fneur.2018.01094. eCollection 2018. Front Neurol. 2018. PMID: 30619053 Free PMC article.
-
Motor and non-motor features in Parkinson's Disease patients carrying GBA gene mutations.Acta Neurol Belg. 2023 Feb;123(1):221-226. doi: 10.1007/s13760-022-02165-y. Epub 2023 Jan 7. Acta Neurol Belg. 2023. PMID: 36609835
References
-
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004;351:1972–1977. - PubMed
-
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010;67:1464–1472. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
