

p19ARF is a critical mediator of both cellular senescence and an innate immune response associated with MYC inactivation in mouse model of acute leukemia
- PMID: 25784651
- PMCID: PMC4414137
- DOI: 10.18632/oncotarget.2969
p19ARF is a critical mediator of both cellular senescence and an innate immune response associated with MYC inactivation in mouse model of acute leukemia
Abstract
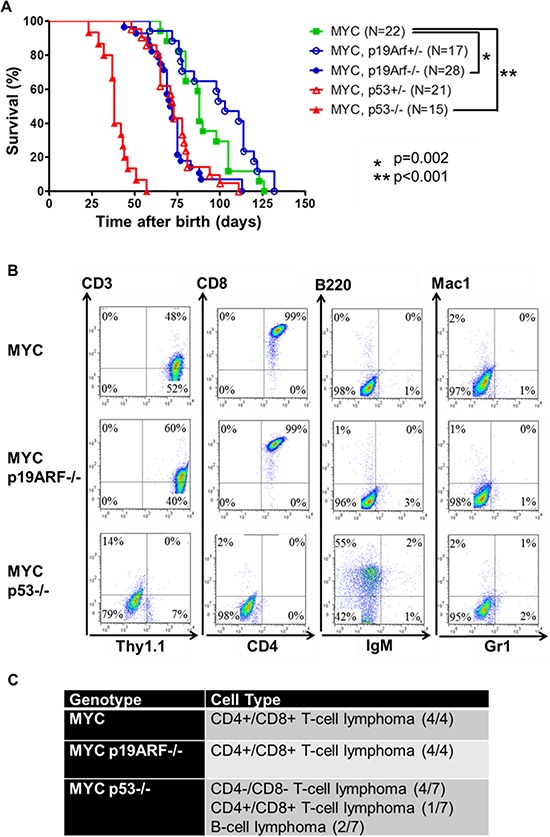
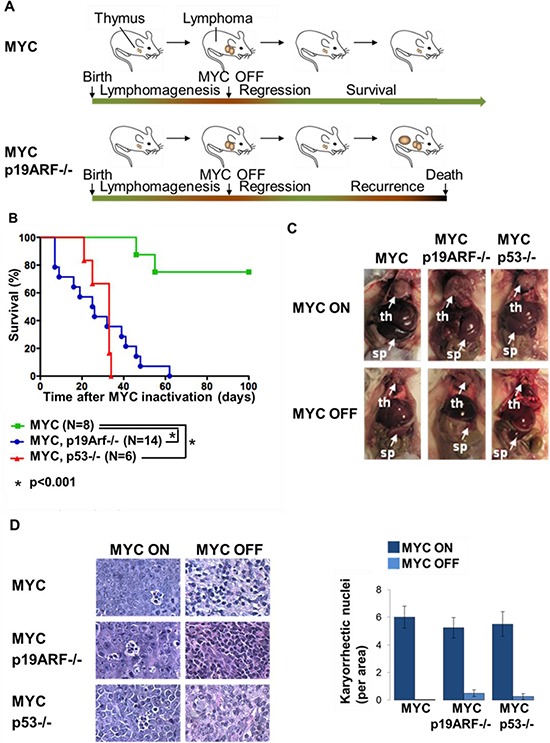
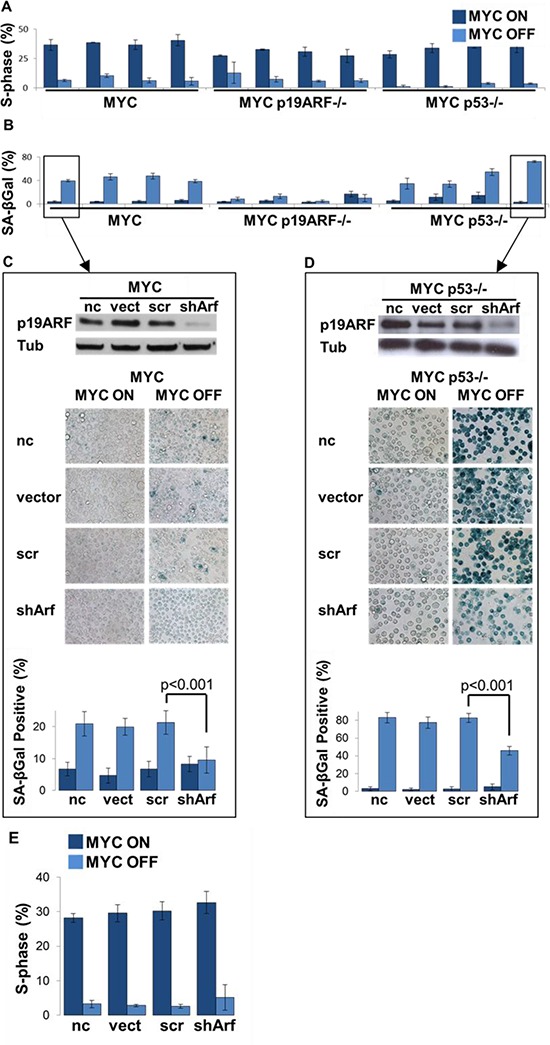
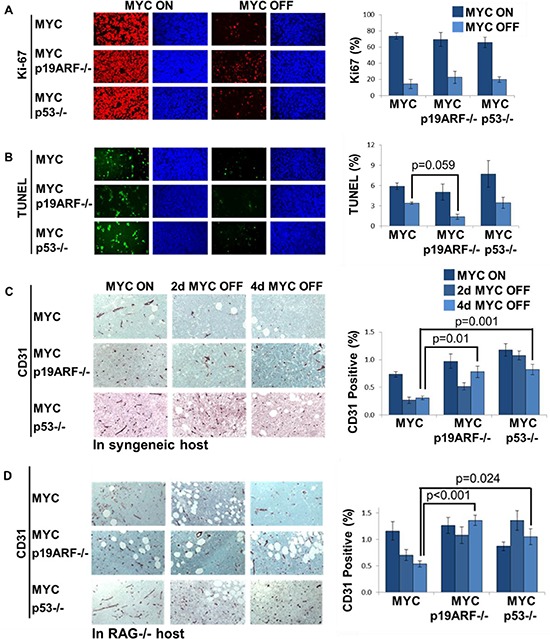
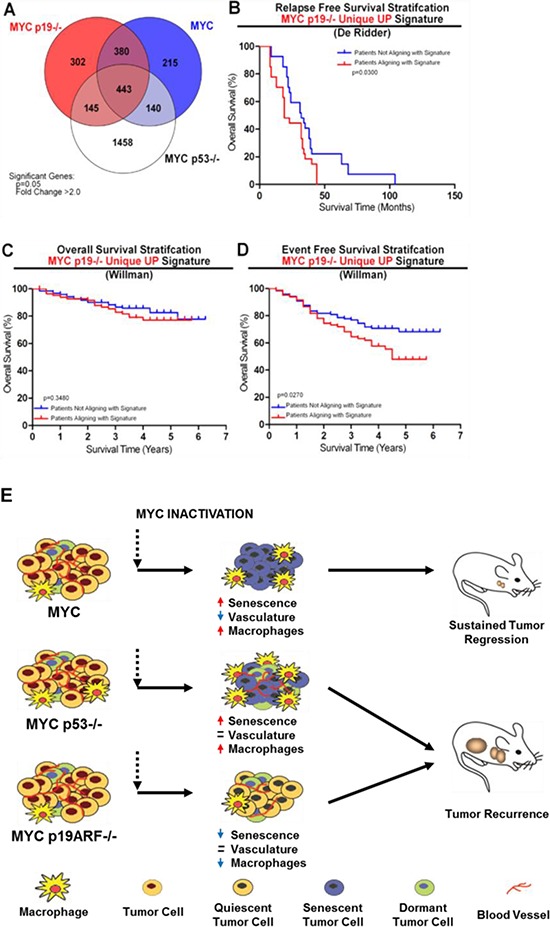
MYC-induced T-ALL exhibit oncogene addiction. Addiction to MYC is a consequence of both cell-autonomous mechanisms, such as proliferative arrest, cellular senescence, and apoptosis, as well as non-cell autonomous mechanisms, such as shutdown of angiogenesis, and recruitment of immune effectors. Here, we show, using transgenic mouse models of MYC-induced T-ALL, that the loss of either p19ARF or p53 abrogates the ability of MYC inactivation to induce sustained tumor regression. Loss of p53 or p19ARF, influenced the ability of MYC inactivation to elicit the shutdown of angiogenesis; however the loss of p19ARF, but not p53, impeded cellular senescence, as measured by SA-beta-galactosidase staining, increased expression of p16INK4A, and specific histone modifications. Moreover, comparative gene expression analysis suggested that a multitude of genes involved in the innate immune response were expressed in p19ARF wild-type, but not null, tumors upon MYC inactivation. Indeed, the loss of p19ARF, but not p53, impeded the in situ recruitment of macrophages to the tumor microenvironment. Finally, p19ARF null-associated gene signature prognosticated relapse-free survival in human patients with ALL. Therefore, p19ARF appears to be important to regulating cellular senescence and innate immune response that may contribute to the therapeutic response of ALL.
Figures


Similar articles
-
Oncogenic c-Myc-induced lymphomagenesis is inhibited non-redundantly by the p19Arf-Mdm2-p53 and RP-Mdm2-p53 pathways.Oncogene. 2015 Nov 12;34(46):5709-17. doi: 10.1038/onc.2015.39. Epub 2015 Mar 30. Oncogene. 2015. PMID: 25823025 Free PMC article.
-
Regulation of the Arf tumor suppressor in Emicro-Myc transgenic mice: longitudinal study of Myc-induced lymphomagenesis.Blood. 2007 Jan 15;109(2):792-4. doi: 10.1182/blood-2006-07-033985. Epub 2006 Sep 12. Blood. 2007. PMID: 16968893
-
Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras.Oncogene. 2000 Mar 23;19(13):1698-709. doi: 10.1038/sj.onc.1203471. Oncogene. 2000. PMID: 10763827
-
MYC activation is a hallmark of cancer initiation and maintenance.Cold Spring Harb Perspect Med. 2014 Jun 2;4(6):a014241. doi: 10.1101/cshperspect.a014241. Cold Spring Harb Perspect Med. 2014. PMID: 24890832 Free PMC article. Review.
-
Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment.Int J Mol Sci. 2016 Nov 19;17(11):1942. doi: 10.3390/ijms17111942. Int J Mol Sci. 2016. PMID: 27869779 Free PMC article. Review.
Cited by
-
Globule Leukocytes and Other Mast Cells in the Mouse Intestine.Vet Pathol. 2018 Jan;55(1):76-97. doi: 10.1177/0300985817705174. Epub 2017 May 11. Vet Pathol. 2018. PMID: 28494703 Free PMC article.
-
Tumor suppressor ARF regulates tissue microenvironment and tumor growth through modulation of macrophage polarization.Oncotarget. 2016 Oct 11;7(41):66835-66850. doi: 10.18632/oncotarget.11652. Oncotarget. 2016. PMID: 27572316 Free PMC article.
-
Loss of p19Arf promotes fibroblast survival during leucine deprivation.Biol Open. 2022 Feb 15;11(2):bio058728. doi: 10.1242/bio.058728. Epub 2022 Feb 17. Biol Open. 2022. PMID: 34994382 Free PMC article.
-
How can same-gene mutations promote both cancer and developmental disorders?Sci Adv. 2022 Jan 14;8(2):eabm2059. doi: 10.1126/sciadv.abm2059. Epub 2022 Jan 14. Sci Adv. 2022. PMID: 35030014 Free PMC article.
-
ARF: connecting senescence and innate immunity for clearance.Aging (Albany NY). 2015 Sep;7(9):613-5. doi: 10.18632/aging.100813. Aging (Albany NY). 2015. PMID: 26412380 Free PMC article.
References
-
- Einsiedel HG, von Stackelberg A, Hartmann R, Fengler R, Schrappe M, Janka-Schaub G, Mann G, Hahlen K, Gobel U, Klingebiel T, Ludwig WD, Henze G. Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Munster Group 87. J Clin Oncol. 2005;23:7942–7950. - PubMed
-
- Erikson J, Finger L, Sun L, ar-Rushdi A, Nishikura K, Minowada J, Finan J, Emanuel BS, Nowell PC, Croce CM. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science. 1986;232:884–886. - PubMed
-
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. - PubMed
-
- Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297:63–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
