

Review
doi: 10.1038/ncb3124.
Epub 2015 Mar 16.
Metabolic pathways promoting cancer cell survival and growth
Affiliations
- PMID: 25774832
- PMCID: PMC4939711
- DOI: 10.1038/ncb3124
Item in Clipboard
Review
Metabolic pathways promoting cancer cell survival and growth
Nat Cell Biol.
2015 Apr.
Abstract
Activation of oncogenes and loss of tumour suppressors promote metabolic reprogramming in cancer, resulting in enhanced nutrient uptake to supply energetic and biosynthetic pathways. However, nutrient limitations within solid tumours may require that malignant cells exhibit metabolic flexibility to sustain growth and survival. Here, we highlight these adaptive mechanisms and also discuss emerging approaches to probe tumour metabolism in vivo and their potential to expand the metabolic repertoire of malignant cells even further.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
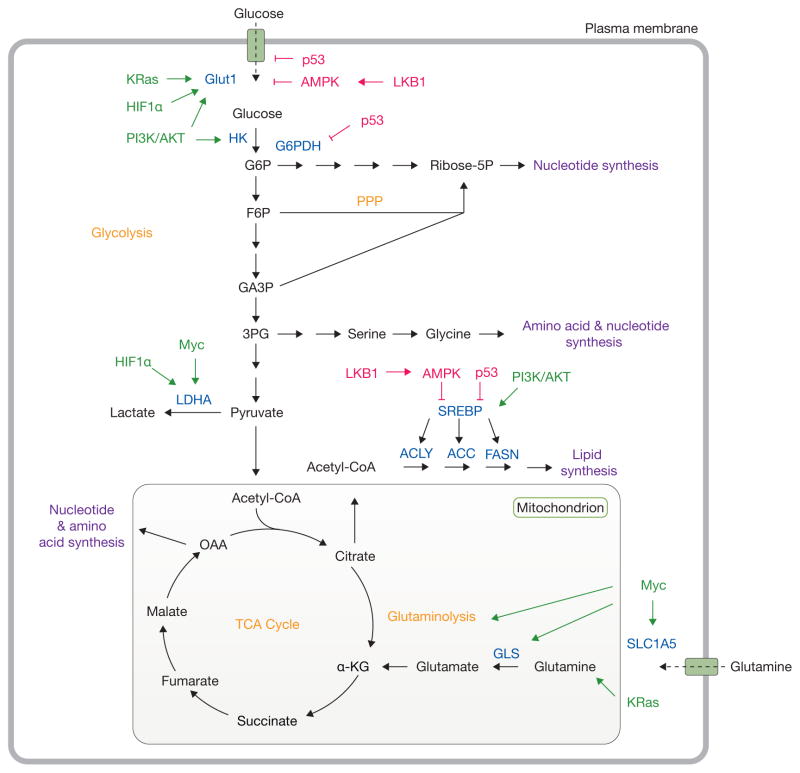
Oncogenic signalling and nutrient availability influence cell metabolism. Oncogenic signalling regulates the acquisition of abundant nutrients including glucose and glutamine, and their utilization to support biosynthetic pathways (purple). Mutations in oncoproteins (green) lead to increased glucose uptake coupled to enhanced lactate production through the Warburg effect. Additionally, KRas and Myc promote glutamine metabolism to fuel the TCA cycle. Intermediates from glycolysis and the TCA cycle supply biosynthetic pathways to produce macromolecules necessary for cell proliferation. Tumour suppressors (red) such as LKB1, AMPK and p53 act at various nodes to oppose biosynthetic metabolism. Important nutrient transporters, transcription factors, and metabolic enzymes are highlighted in blue. Classic metabolic pathways are highlighted in orange. HK, hexokinase; G6P, glucose-6-phosphate; G6PDH, glucose-6-phosphate dehydrogenase; Ribose-5P, ribose-5-phosphate; F6P, fructose-6-phosphate; PPP, pentose phosphate pathway; GA3P, glyceraldehyde-3-phosphate; 3PG, 3-phosphoglycerate; LDHA, lactate dehydrogenase A; SREBP, sterol regulatory element-binding protein; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; GLS, glutaminase, α-KG, alpha-ketoglutarate; OAA, oxaloacetate; TCA, tricarboxylic acid.
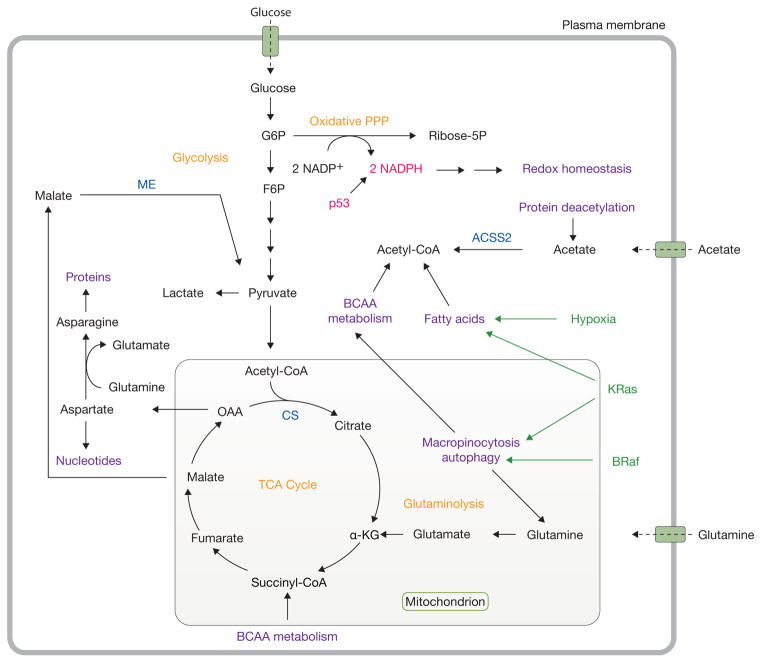
Catabolic pathways support metabolism during nutrient stress. Under conditions of nutrient deprivation and other stresses, tumorigenic mutations also reprogram metabolism to support cell survival. In particular, KRas stimulates autophagy and macropinocytosis and facilitates fatty acid uptake. Mutations in BRaf also lead to enhanced autophagy. p53 promotes NADPH production through the pentose phosphate pathway to maintain redox homeostasis. Acetate and branched-chain amino acids serve as alternative substrates to support metabolism. Oncogenic drivers are highlighted in green, while tumour suppressors are shown in red. Important metabolic enzymes are highlighted in blue. Classic cancer metabolic pathways are shown in orange, while emerging pathways and activities supporting cell proliferation are shown in purple. G6P, glucose-6-phosphate; PPP, pentose phosphate pathway; ribose-5P, ribose-5-phosphate; F6P, fructose-6-phosphate; TCA, tricarboxylic acid; α-KG, alpha-ketoglutarate; OAA, oxaloacetate; BCAA, branched-chain amino acid; CS, citrate synthase; ME, malic enzyme; ACSS2, acetyl-CoA synthetase 2.
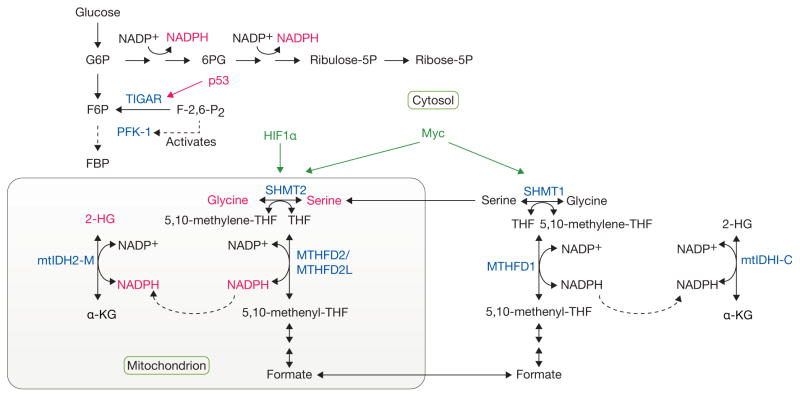
Complex, compartmentalized pathways of NADPH production. Metabolic tracing experiments have provided tools to follow the cytosolic production of NADPH through the pentose phosphate pathway (PPP), as well as the contribution of serine/glycine metabolism to NADPH generation in the mitochondria. Given that mutant forms of isocitrate dehydrogenase (mtIDH1/2), localized to the cytosol (C) or mitochondria (M), respectively, give rise to increased 2-hydroxyglutarate (2-HG) levels, this reporter system was used to trace the production of NADPH in the mitochondria from serine/glycine through methylenetetrahydrofolate dehydrogenase 2 (like) (MTHFD2/MTHFD2L) enzymes. p53 regulates NADPH production by inducing the expression of TIGAR, a fructose-2,6-bisphosphatase, which reduces the levels of fructose-2,6-bisphosphate (F-2,6-P2), resulting in reduced activation of phosphofructokinase-1 (PFK-1). This reduction in the rate of glycolysis, allows for shunting of glycolytic intermediates into the PPP and the concomitant generation of NADPH. HIF1α and Myc regulate the expression of serine hydroxymethyltransferase (SHMT1/2) enzymes, thereby regulating serine/glycine metabolism. Tumor suppressors and oncogenes are shown in red and green, respectively. Important metabolic enzymes involved in these pathways are shown in blue. G6P, glucose-6-phosphate; 6PG, 6-phosphogluconate; Ribulose-5P, ribulose-5-phosphate; Ribose-5P, ribose-5-phosphate; F6P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; THF, tetrahydrofolate; α-KG, alpha-ketoglutarate.
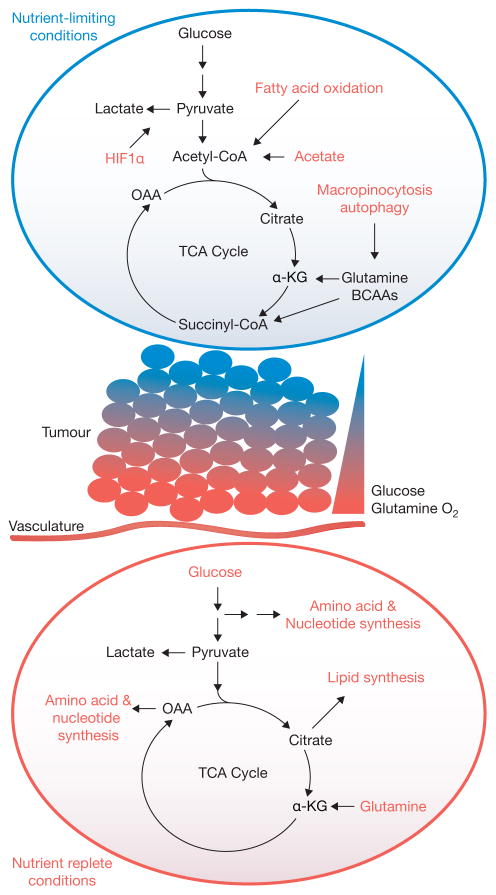
A model for metabolic heterogeneity in solid tumours. Within solid tumours, regional metabolic activities likely vary significantly according to influences of the microenvironment, particularly access to nutrients and oxygen. In the illustrated example, cells located close to vasculature use their favourable access to nutrients and oxygen to feed oncogene-stimulated anabolic pathways. However, gradients of nutrient availability demand that alternative pathways, including macromolecular degradation, are increasingly engaged at more remote sites to support cell viability. Oncogenes may also enhance the cell’s ability to activate these pathways. OAA, oxaloacetate; α-KG, alpha-ketoglutarate; TCA, tricarboxylic acid; BCAA, branched-chain amino acid.
Similar articles
-
Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells.Int J Biochem Cell Biol. 2011 Jul;43(7):950-68. doi: 10.1016/j.biocel.2010.05.003. Epub 2010 May 10. Int J Biochem Cell Biol. 2011. PMID: 20460169 Review.
-
The Role of Glucose and Lipid Metabolism in Growth and Survival of Cancer Cells.Recent Results Cancer Res. 2016;207:1-22. doi: 10.1007/978-3-319-42118-6_1. Recent Results Cancer Res. 2016. PMID: 27557532 Review.
-
Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis.Semin Cell Dev Biol. 2012 Jun;23(4):362-9. doi: 10.1016/j.semcdb.2012.02.002. Epub 2012 Feb 11. Semin Cell Dev Biol. 2012. PMID: 22349059 Review.
-
Metabolic transformation in cancer.Carcinogenesis. 2009 Aug;30(8):1269-80. doi: 10.1093/carcin/bgp070. Epub 2009 Mar 25. Carcinogenesis. 2009. PMID: 19321800 Review.
-
Glucose avidity of carcinomas.Cancer Lett. 2009 Apr 18;276(2):125-35. doi: 10.1016/j.canlet.2008.08.007. Epub 2008 Sep 14. Cancer Lett. 2009. PMID: 18790562 Review.
Cited by
-
Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN.Int J Mol Sci. 2023 Nov 19;24(22):16503. doi: 10.3390/ijms242216503. Int J Mol Sci. 2023. PMID: 38003694 Free PMC article.
-
How DNA methylation affects the Warburg effect.Int J Biol Sci. 2020 Apr 27;16(12):2029-2041. doi: 10.7150/ijbs.45420. eCollection 2020. Int J Biol Sci. 2020. PMID: 32549751 Free PMC article. Review.
-
Enzymes for N-Glycan Branching and Their Genetic and Nongenetic Regulation in Cancer.Biomolecules. 2016 Apr 28;6(2):25. doi: 10.3390/biom6020025. Biomolecules. 2016. PMID: 27136596 Free PMC article. Review.
-
PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development.Cell Rep. 2016 Aug 30;16(9):2415-27. doi: 10.1016/j.celrep.2016.07.082. Epub 2016 Aug 18. Cell Rep. 2016. PMID: 27545895 Free PMC article.
-
The Role of Tumor Metabolic Reprogramming in Tumor Immunity.Int J Mol Sci. 2023 Dec 13;24(24):17422. doi: 10.3390/ijms242417422. Int J Mol Sci. 2023. PMID: 38139250 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
