

Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain
- PMID: 25744681
- PMCID: PMC4664460
- DOI: 10.1016/bs.pmbts.2014.11.010
Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain
Abstract
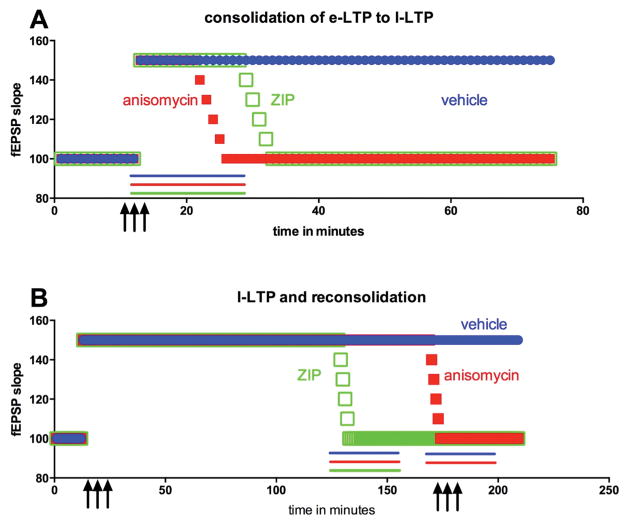
Pain sensing neurons in the periphery (called nociceptors) and the central neurons that receive their projections show remarkable plasticity following injury. This plasticity results in amplification of pain signaling that is now understood to be crucial for the recovery and survival of organisms following injury. These same plasticity mechanisms may drive a transition to a nonadaptive chronic pain state if they fail to resolve following the termination of the healing process. Remarkable advances have been achieved in the past two decades in understanding the molecular mechanisms that underlie pain plasticity following injury. The mechanisms bear a striking resemblance to molecular mechanisms involved in learning and memory processes in other brain regions, including the hippocampus and cerebral cortex. Here those mechanisms, their commonalities and subtle differences, will be highlighted and their role in causing chronic pain will be discussed. Arising from these data is the striking argument that chronic pain is a disease of the nervous system, which distinguishes this phenomena from acute pain that is frequently a symptom alerting the organism to injury. This argument has important implications for the development of disease modifying therapeutics.
Keywords: AMPK; Atypical PKC; Axonal mRNA; BDNF; CREB; ERK; Hyperalgesic priming; IL-6; LTP; NGF; Opioid; PKMzeta; Reconsolidation; eIF4E; mTOR; trkB.
© 2015 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
The pharmacology of nociceptor priming.Handb Exp Pharmacol. 2015;227:15-37. doi: 10.1007/978-3-662-46450-2_2. Handb Exp Pharmacol. 2015. PMID: 25846612 Free PMC article. Review.
-
ZIPping to pain relief: the role (or not) of PKMζ in chronic pain.Mol Pain. 2013 Feb 22;9:6. doi: 10.1186/1744-8069-9-6. Mol Pain. 2013. PMID: 23433248 Free PMC article. Review.
-
eIF4E Phosphorylation Influences Bdnf mRNA Translation in Mouse Dorsal Root Ganglion Neurons.Front Cell Neurosci. 2018 Feb 6;12:29. doi: 10.3389/fncel.2018.00029. eCollection 2018. Front Cell Neurosci. 2018. PMID: 29467623 Free PMC article.
-
Roles of Proton-Sensing Receptors in the Transition from Acute to Chronic Pain.J Dent Res. 2016 Feb;95(2):135-42. doi: 10.1177/0022034515618382. Epub 2015 Nov 23. J Dent Res. 2016. PMID: 26597969 Review.
-
eIF4E phosphorylation regulates ongoing pain, independently of inflammation, and hyperalgesic priming in the mouse CFA model.Neurobiol Pain. 2018 Aug-Dec;4:45-50. doi: 10.1016/j.ynpai.2018.03.001. Epub 2018 Mar 15. Neurobiol Pain. 2018. PMID: 30211343 Free PMC article.
Cited by
-
Systems Biology Guided Gene Enrichment Approaches Improve Prediction of Chronic Post-surgical Pain After Spine Fusion.Front Genet. 2021 Mar 23;12:594250. doi: 10.3389/fgene.2021.594250. eCollection 2021. Front Genet. 2021. PMID: 33868360 Free PMC article.
-
Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism.J Neurosci. 2015 Apr 22;35(16):6307-17. doi: 10.1523/JNEUROSCI.3481-14.2015. J Neurosci. 2015. PMID: 25904784 Free PMC article.
-
Nociceptive Biology of Molluscs and Arthropods: Evolutionary Clues About Functions and Mechanisms Potentially Related to Pain.Front Physiol. 2018 Aug 3;9:1049. doi: 10.3389/fphys.2018.01049. eCollection 2018. Front Physiol. 2018. PMID: 30123137 Free PMC article. Review.
-
Chronic Daily Headache: Mechanisms and Principles of Management.Curr Pain Headache Rep. 2016 Feb;20(2):10. doi: 10.1007/s11916-016-0542-3. Curr Pain Headache Rep. 2016. PMID: 26780038 Review.
-
Inhibition of Mammalian Target of Rapamycin (mTOR) Signaling in the Insular Cortex Alleviates Neuropathic Pain after Peripheral Nerve Injury.Front Mol Neurosci. 2017 Mar 21;10:79. doi: 10.3389/fnmol.2017.00079. eCollection 2017. Front Mol Neurosci. 2017. PMID: 28377693 Free PMC article.
References
-
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem. 2008a;89:260–268. - PubMed
-
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiology of learning and memory. 2008b;89:260–268. - PubMed
-
- Althaus A, Hinrichs-Rocker A, Chapman R, Arranz Becker O, Lefering R, Simanski C, Weber F, Moser KH, Joppich R, Trojan S, Gutzeit N, Neugebauer E. Development of a risk index for the prediction of chronic post-surgical pain. Eur J Pain. 2012;16:901–910. - PubMed
-
- Andersson GB. Epidemiological features of chronic low-back pain. Lancet. 1999;354:581–585. - PubMed
-
- Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F, Price TJ. Spinal protein kinase M zeta underlies the maintenance mechanism of persistent nociceptive sensitization. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:6646–6653. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
