

Origins and Evolutionary Dynamics of H3N2 Canine Influenza Virus
- PMID: 25740996
- PMCID: PMC4442499
- DOI: 10.1128/JVI.03395-14
Origins and Evolutionary Dynamics of H3N2 Canine Influenza Virus
Abstract
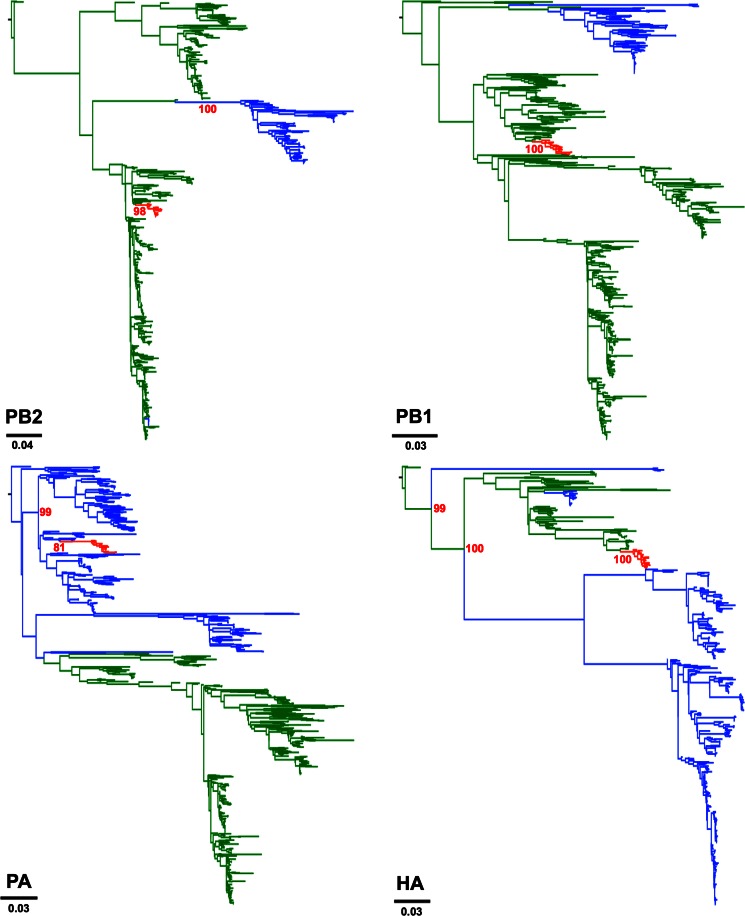
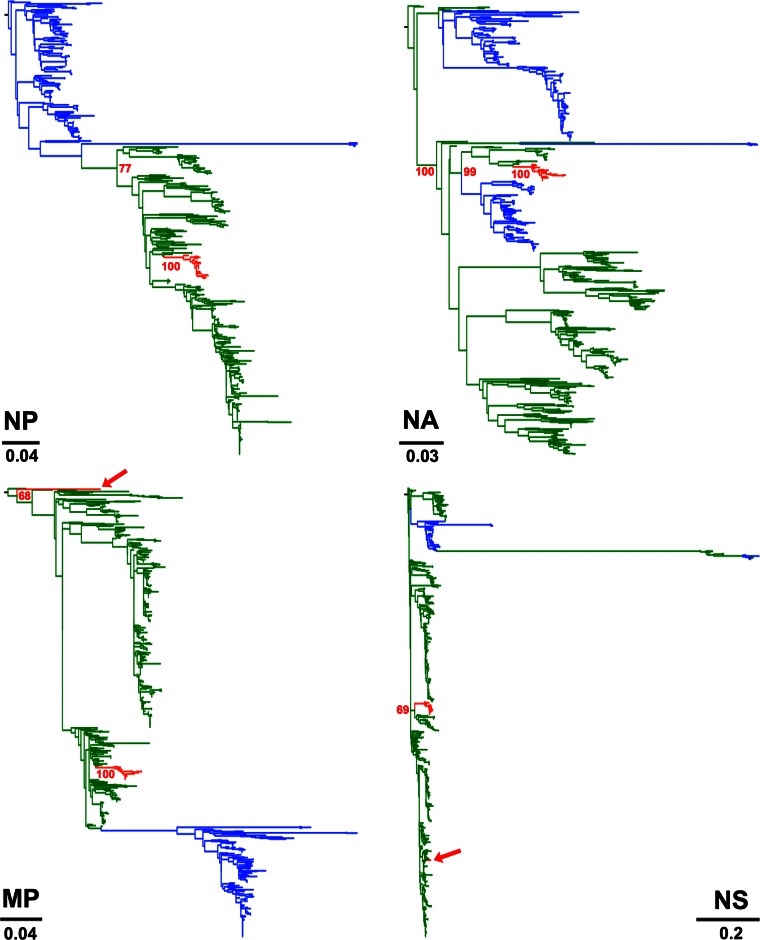
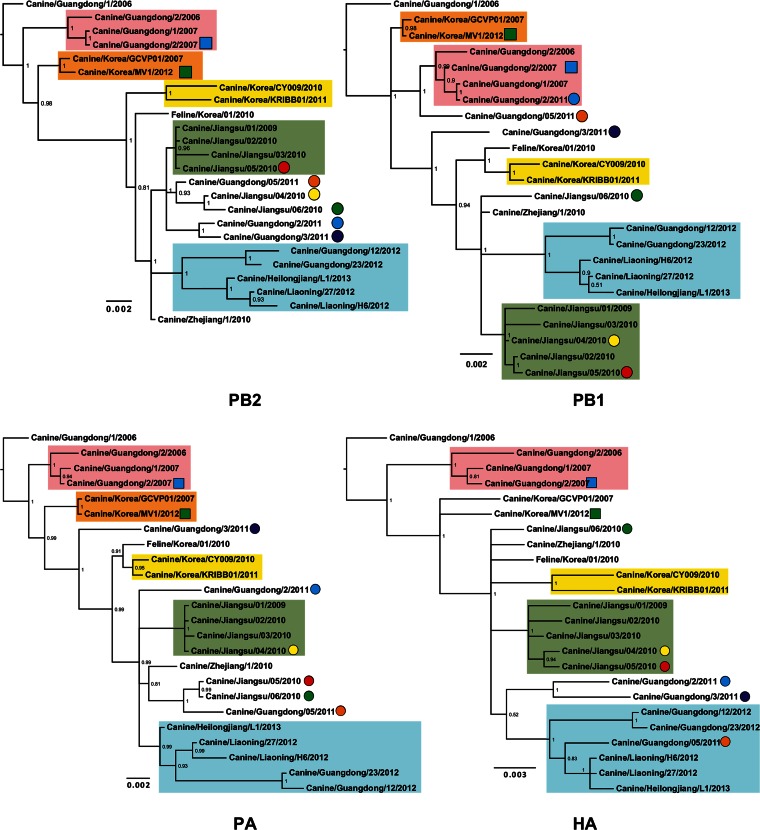
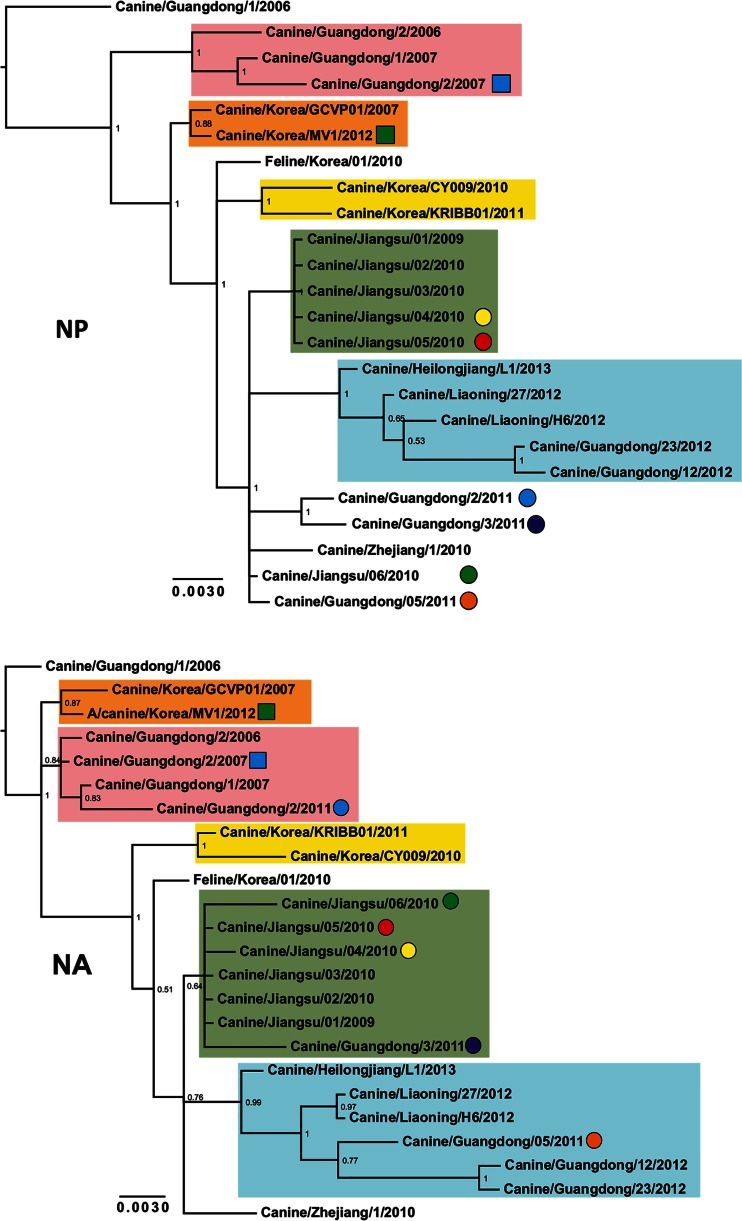
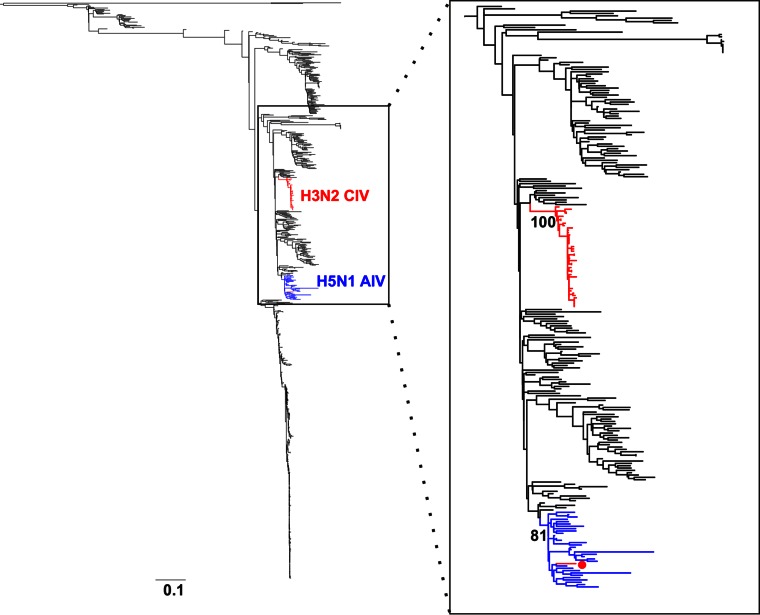
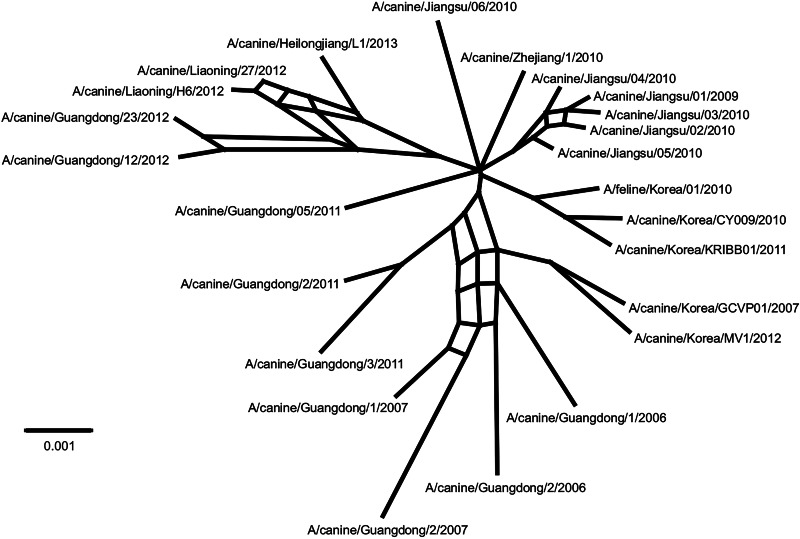
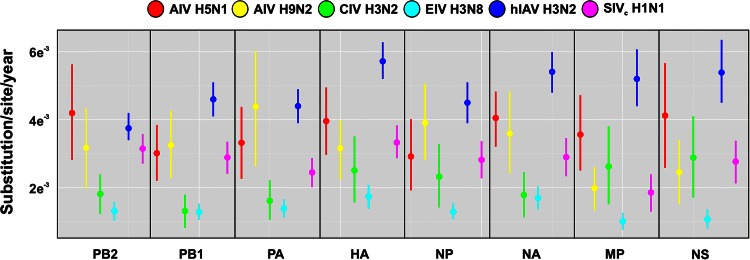
Influenza A viruses (IAVs) are maintained mainly in wild birds, and despite frequent spillover infections of avian IAVs into mammals, only a small number of viruses have become established in mammalian hosts. A new H3N2 canine influenza virus (CIV) of avian origin emerged in Asia in the mid-2000s and is now circulating in dog populations of China and South Korea, and possibly in Thailand. The emergence of CIV provides new opportunities for zoonotic infections and interspecies transmission. We examined 14,764 complete IAV genomes together with all CIV genomes publicly available since its first isolation until 2013. We show that CIV may have originated as early as 1999 as a result of segment reassortment among Eurasian and North American avian IAV lineages. We also identified amino acid changes that might have played a role in CIV emergence, some of which have not been previously identified in other cross-species jumps. CIV evolves at a lower rate than H3N2 human influenza viruses do, and viral phylogenies exhibit geographical structure compatible with high levels of local transmission. We detected multiple intrasubtypic and heterosubtypic reassortment events, including the acquisition of the NS segment of an H5N1 avian influenza virus that had previously been overlooked. In sum, our results provide insight into the adaptive changes required by avian viruses to establish themselves in mammals and also highlight the potential role of dogs to act as intermediate hosts in which viruses with zoonotic and/or pandemic potential could originate, particularly with an estimated dog population of ∼ 700 million.
Importance: Influenza A viruses circulate in humans and animals. This multihost ecology has important implications, as past pandemics were caused by IAVs carrying gene segments of both human and animal origin. Adaptive evolution is central to cross-species jumps, and this is why understanding the evolutionary processes that shape influenza A virus genomes is key to elucidating the mechanisms underpinning viral emergence. An avian-origin canine influenza virus (CIV) has recently emerged in dogs and is spreading in Asia. We reconstructed the evolutionary history of CIV and show that it originated from both Eurasian and North American avian lineages. We also identified the mutations that might have been responsible for the cross-species jump. Finally, we provide evidence of multiple reassortment events between CIV and other influenza viruses (including an H5N1 avian virus). This is a cause for concern, as there is a large global dog population to which humans are highly exposed.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
The ecology and adaptive evolution of influenza A interspecies transmission.Influenza Other Respir Viruses. 2017 Jan;11(1):74-84. doi: 10.1111/irv.12412. Epub 2016 Aug 8. Influenza Other Respir Viruses. 2017. PMID: 27426214 Free PMC article. Review.
-
Emergence and Evolution of Novel Reassortant Influenza A Viruses in Canines in Southern China.mBio. 2018 Jun 5;9(3):e00909-18. doi: 10.1128/mBio.00909-18. mBio. 2018. PMID: 29871917 Free PMC article.
-
Emergence and adaptation of H3N2 canine influenza virus from avian influenza virus: An overlooked role of dogs in interspecies transmission.Transbound Emerg Dis. 2019 Mar;66(2):842-851. doi: 10.1111/tbed.13093. Epub 2019 Jan 5. Transbound Emerg Dis. 2019. PMID: 30520554
-
Zoonotic Risk, Pathogenesis, and Transmission of Avian-Origin H3N2 Canine Influenza Virus.J Virol. 2017 Oct 13;91(21):e00637-17. doi: 10.1128/JVI.00637-17. Print 2017 Nov 1. J Virol. 2017. PMID: 28814512 Free PMC article.
-
[Swine influenza virus: evolution mechanism and epidemic characterization--a review].Wei Sheng Wu Xue Bao. 2009 Sep;49(9):1138-45. Wei Sheng Wu Xue Bao. 2009. PMID: 20030049 Review. Chinese.
Cited by
-
The C-terminal amino acid motifs of NS1 protein affect the replication and virulence of naturally NS-truncated H1N1 canine influenza virus.Emerg Microbes Infect. 2024 Dec;13(1):2400546. doi: 10.1080/22221751.2024.2400546. Epub 2024 Sep 13. Emerg Microbes Infect. 2024. PMID: 39221898 Free PMC article.
-
The ecology and adaptive evolution of influenza A interspecies transmission.Influenza Other Respir Viruses. 2017 Jan;11(1):74-84. doi: 10.1111/irv.12412. Epub 2016 Aug 8. Influenza Other Respir Viruses. 2017. PMID: 27426214 Free PMC article. Review.
-
Molecular surveillance of traditional and emerging pathogens associated with canine infectious respiratory disease.Vet Microbiol. 2016 Aug 30;192:21-25. doi: 10.1016/j.vetmic.2016.06.009. Epub 2016 Jun 22. Vet Microbiol. 2016. PMID: 27527760 Free PMC article.
-
Multiple Incursions and Recurrent Epidemic Fade-Out of H3N2 Canine Influenza A Virus in the United States.J Virol. 2018 Jul 31;92(16):e00323-18. doi: 10.1128/JVI.00323-18. Print 2018 Aug 15. J Virol. 2018. PMID: 29875234 Free PMC article.
-
Molecular analyses of H3N2 canine influenza viruses isolated from Korea during 2013-2014.Virus Genes. 2016 Apr;52(2):204-17. doi: 10.1007/s11262-015-1274-x. Epub 2016 Jan 25. Virus Genes. 2016. PMID: 26810402 Free PMC article.
References
-
- Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. 2012. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog 8:e1002998. doi:10.1371/journal.ppat.1002998. - DOI - PMC - PubMed
-
- Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New world bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657. doi:10.1371/journal.ppat.1003657. - DOI - PMC - PubMed
-
- Waddell GH, Teigland MB, Sigel MM. 1963. A new influenza virus associated with equine respiratory disease. J Am Vet Med Assoc 143:587–590. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
