

Structural basis for self-assembly of a cytolytic pore lined by protein and lipid
- PMID: 25716479
- PMCID: PMC4351601
- DOI: 10.1038/ncomms7337
Structural basis for self-assembly of a cytolytic pore lined by protein and lipid
Abstract
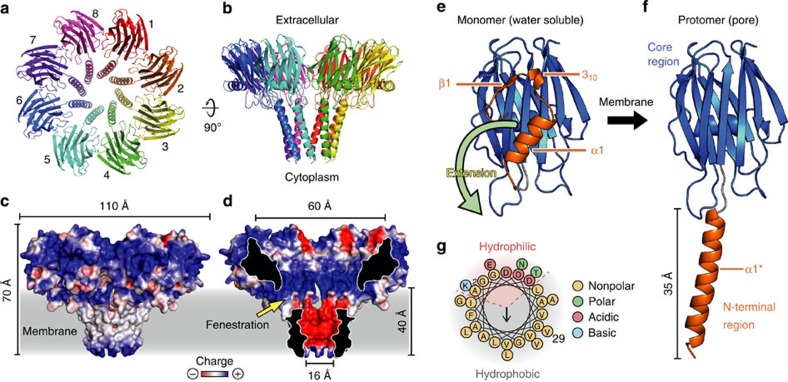
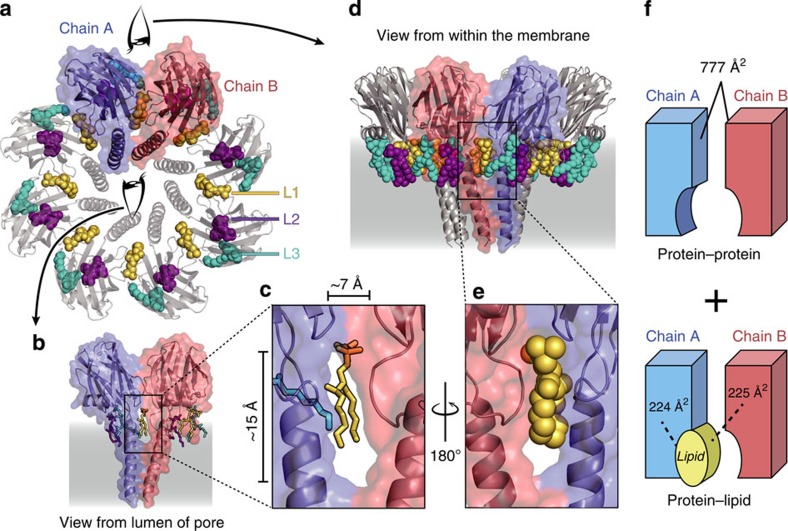
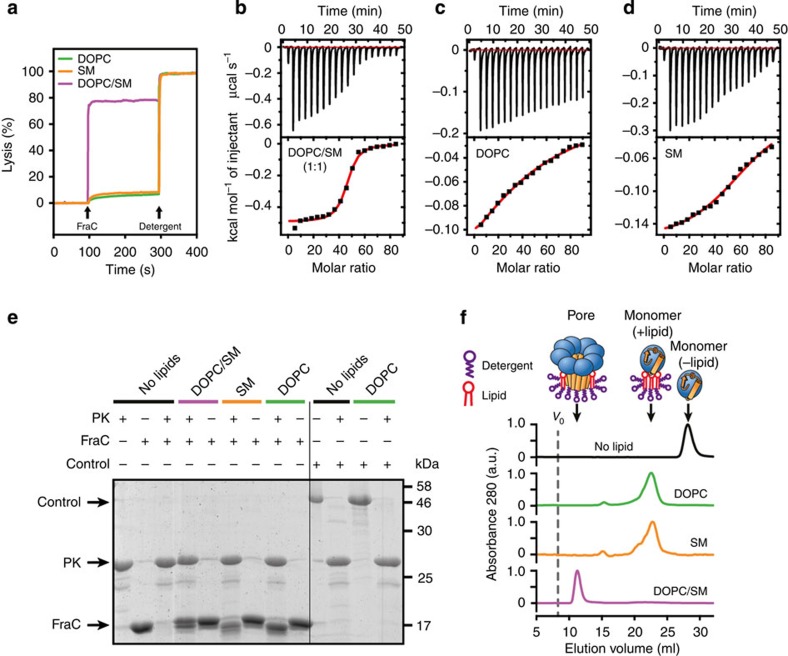
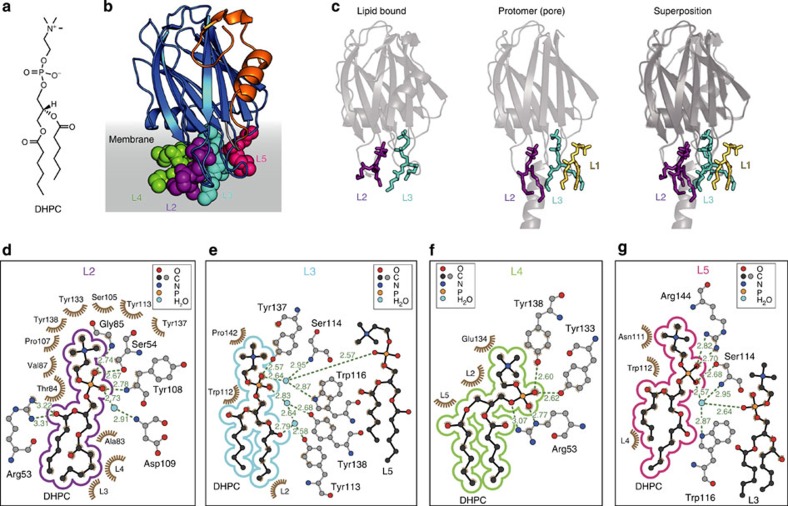
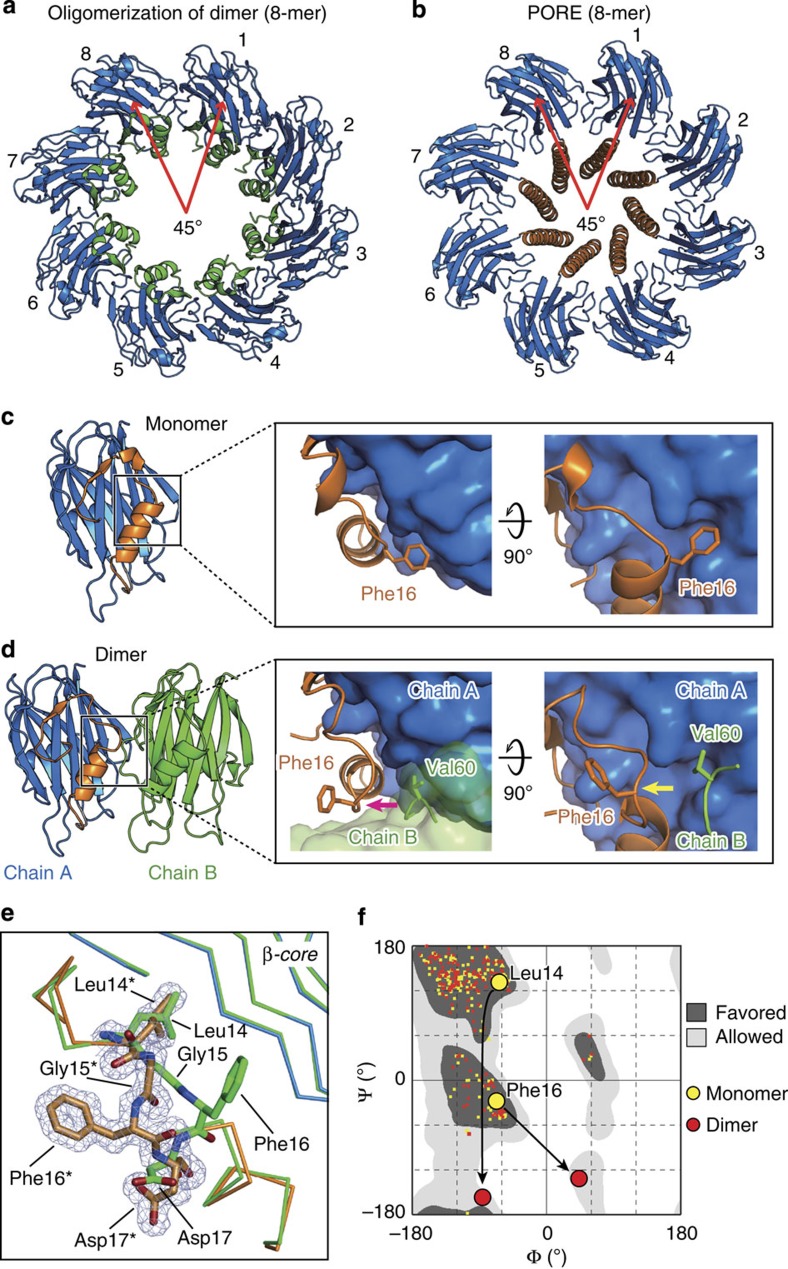
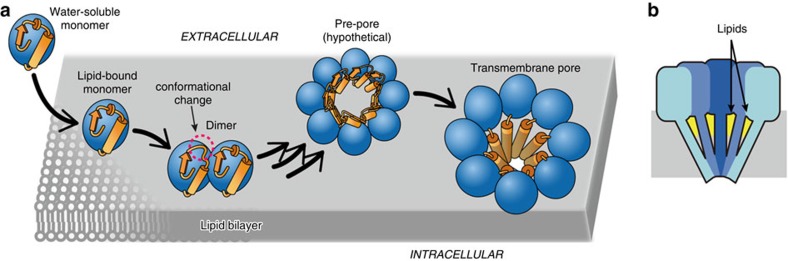
Pore-forming toxins (PFT) are water-soluble proteins that possess the remarkable ability to self-assemble on the membrane of target cells, where they form pores causing cell damage. Here, we elucidate the mechanism of action of the haemolytic protein fragaceatoxin C (FraC), a α-barrel PFT, by determining the crystal structures of FraC at four different stages of the lytic mechanism, namely the water-soluble state, the monomeric lipid-bound form, an assembly intermediate and the fully assembled transmembrane pore. The structure of the transmembrane pore exhibits a unique architecture composed of both protein and lipids, with some of the lipids lining the pore wall, acting as assembly cofactors. The pore also exhibits lateral fenestrations that expose the hydrophobic core of the membrane to the aqueous environment. The incorporation of lipids from the target membrane within the structure of the pore provides a membrane-specific trigger for the activation of a haemolytic toxin.
Figures
Similar articles
-
A pore-forming toxin requires a specific residue for its activity in membranes with particular physicochemical properties.J Biol Chem. 2015 Apr 24;290(17):10850-61. doi: 10.1074/jbc.M114.615211. Epub 2015 Mar 10. J Biol Chem. 2015. PMID: 25759390 Free PMC article.
-
Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins.Structure. 2011 Feb 9;19(2):181-91. doi: 10.1016/j.str.2010.11.013. Structure. 2011. PMID: 21300287
-
Bidirectional Transformation of a Metamorphic Protein between the Water-Soluble and Transmembrane Native States.Biochemistry. 2015 Nov 24;54(46):6863-6. doi: 10.1021/acs.biochem.5b01112. Epub 2015 Nov 11. Biochemistry. 2015. PMID: 26544760
-
Pore formation by actinoporins, cytolysins from sea anemones.Biochim Biophys Acta. 2016 Mar;1858(3):446-56. doi: 10.1016/j.bbamem.2015.09.007. Epub 2015 Sep 6. Biochim Biophys Acta. 2016. PMID: 26351738 Review.
-
The multigene families of actinoporins (part II): Strategies for heterologous production in Escherichia coli.Toxicon. 2016 Aug;118:64-81. doi: 10.1016/j.toxicon.2016.03.018. Epub 2016 Apr 11. Toxicon. 2016. PMID: 27080349 Review.
Cited by
-
The Manipulation of the Internal Hydrophobicity of FraC Nanopores Augments Peptide Capture and Recognition.ACS Nano. 2021 Jun 22;15(6):9600-9613. doi: 10.1021/acsnano.0c09958. Epub 2021 Jun 1. ACS Nano. 2021. PMID: 34060809 Free PMC article.
-
Reversible Photocontrolled Nanopore Assembly.J Am Chem Soc. 2019 Sep 11;141(36):14356-14363. doi: 10.1021/jacs.9b06998. Epub 2019 Aug 30. J Am Chem Soc. 2019. PMID: 31469268 Free PMC article.
-
Identification of a pore-forming protein from sea anemone Anthopleura dowii Verrill (1869) venom by mass spectrometry.J Venom Anim Toxins Incl Trop Dis. 2019 Feb 11;25:e147418. doi: 10.1590/1678-9199-JVATITD-1474-18. eCollection 2019. J Venom Anim Toxins Incl Trop Dis. 2019. PMID: 31131002 Free PMC article.
-
Functional and Structural Variation among Sticholysins, Pore-Forming Proteins from the Sea Anemone Stichodactyla helianthus.Int J Mol Sci. 2020 Nov 24;21(23):8915. doi: 10.3390/ijms21238915. Int J Mol Sci. 2020. PMID: 33255441 Free PMC article. Review.
-
Martini 3: a general purpose force field for coarse-grained molecular dynamics.Nat Methods. 2021 Apr;18(4):382-388. doi: 10.1038/s41592-021-01098-3. Epub 2021 Mar 29. Nat Methods. 2021. PMID: 33782607
References
-
- Anderluh G. & Lakey J. H. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33, 482–490 (2008). - PubMed
-
- Rosado C. J. et al.. A common fold mediates vertebrate defense and bacterial attack. Science 317, 1548–1551 (2007). - PubMed
-
- Iacovache I., van der Goot F. G. & Pernot L. Pore formation: an ancient yet complex form of attack. Biochim. Biophys. Acta 1778, 1611–1623 (2008). - PubMed
-
- Geny B. & Popoff M. R. Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol. Cell 98, 667–678 (2006). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
