

Protein models docking benchmark 2
- PMID: 25712716
- PMCID: PMC4400263
- DOI: 10.1002/prot.24784
Protein models docking benchmark 2
Abstract
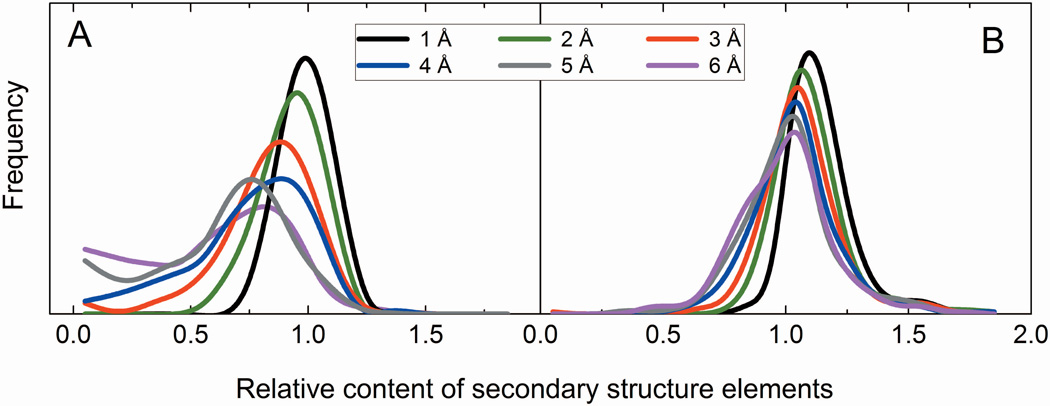
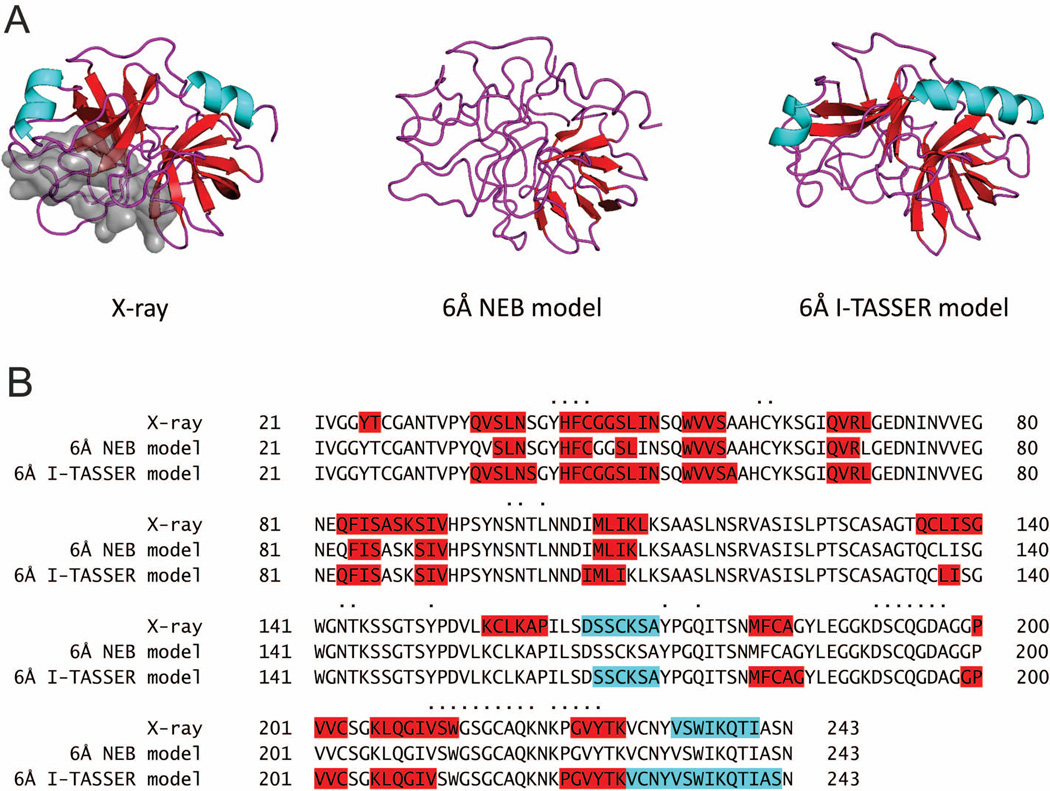
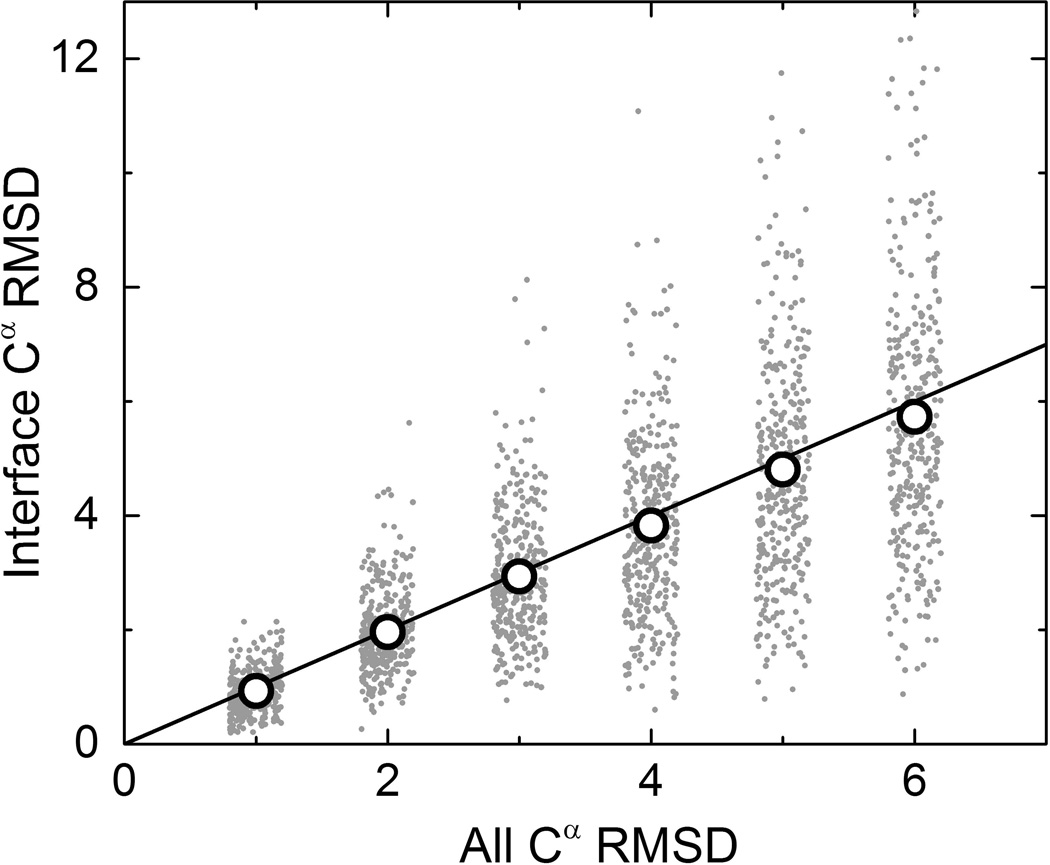
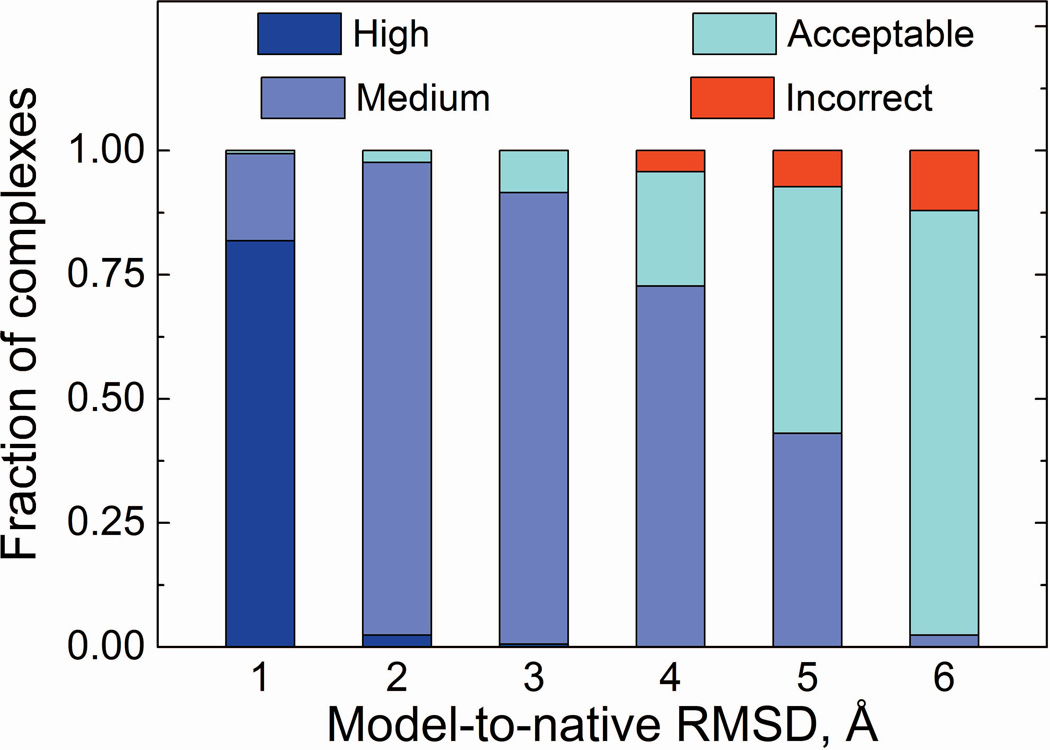
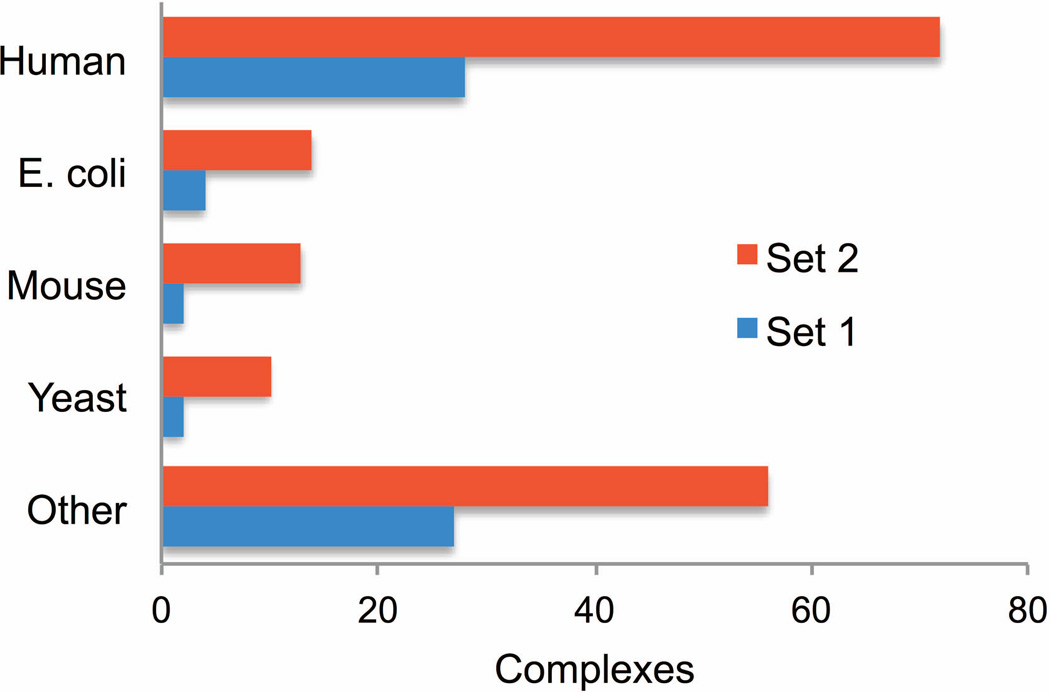
Structural characterization of protein-protein interactions is essential for our ability to understand life processes. However, only a fraction of known proteins have experimentally determined structures. Such structures provide templates for modeling of a large part of the proteome, where individual proteins can be docked by template-free or template-based techniques. Still, the sensitivity of the docking methods to the inherent inaccuracies of protein models, as opposed to the experimentally determined high-resolution structures, remains largely untested, primarily due to the absence of appropriate benchmark set(s). Structures in such a set should have predefined inaccuracy levels and, at the same time, resemble actual protein models in terms of structural motifs/packing. The set should also be large enough to ensure statistical reliability of the benchmarking results. We present a major update of the previously developed benchmark set of protein models. For each interactor, six models were generated with the model-to-native C(α) RMSD in the 1 to 6 Å range. The models in the set were generated by a new approach, which corresponds to the actual modeling of new protein structures in the "real case scenario," as opposed to the previous set, where a significant number of structures were model-like only. In addition, the larger number of complexes (165 vs. 63 in the previous set) increases the statistical reliability of the benchmarking. We estimated the highest accuracy of the predicted complexes (according to CAPRI criteria), which can be attained using the benchmark structures. The set is available at http://dockground.bioinformatics.ku.edu.
Keywords: modeling of protein complexes; protein interactions; protein modeling; protein recognition; structure prediction.
© 2015 Wiley Periodicals, Inc.
Figures


Similar articles
-
Protein models: the Grand Challenge of protein docking.Proteins. 2014 Feb;82(2):278-87. doi: 10.1002/prot.24385. Epub 2013 Oct 17. Proteins. 2014. PMID: 23934791 Free PMC article.
-
Structural templates for comparative protein docking.Proteins. 2015 Sep;83(9):1563-70. doi: 10.1002/prot.24736. Epub 2015 Jun 13. Proteins. 2015. PMID: 25488330 Free PMC article.
-
Simulated unbound structures for benchmarking of protein docking in the DOCKGROUND resource.BMC Bioinformatics. 2015 Jul 31;16(1):243. doi: 10.1186/s12859-015-0672-3. BMC Bioinformatics. 2015. PMID: 26227548 Free PMC article.
-
Advances in template-based protein docking by utilizing interfaces towards completing structural interactome.Curr Opin Struct Biol. 2015 Dec;35:87-92. doi: 10.1016/j.sbi.2015.10.001. Epub 2015 Nov 9. Curr Opin Struct Biol. 2015. PMID: 26539658 Review.
-
Low-resolution structural modeling of protein interactome.Curr Opin Struct Biol. 2013 Apr;23(2):198-205. doi: 10.1016/j.sbi.2012.12.003. Epub 2013 Jan 5. Curr Opin Struct Biol. 2013. PMID: 23294579 Free PMC article. Review.
Cited by
-
Surfing the Protein-Protein Interaction Surface Using Docking Methods: Application to the Design of PPI Inhibitors.Molecules. 2015 Jun 23;20(6):11569-603. doi: 10.3390/molecules200611569. Molecules. 2015. PMID: 26111183 Free PMC article. Review.
-
A benchmark testing ground for integrating homology modeling and protein docking.Proteins. 2017 Jan;85(1):10-16. doi: 10.1002/prot.25063. Epub 2016 Nov 13. Proteins. 2017. PMID: 27172383 Free PMC article.
-
Pushing the Backbone in Protein-Protein Docking.Structure. 2016 Oct 4;24(10):1821-1829. doi: 10.1016/j.str.2016.06.025. Epub 2016 Aug 25. Structure. 2016. PMID: 27568930 Free PMC article.
-
Biological function derived from predicted structures in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):370-91. doi: 10.1002/prot.24997. Epub 2016 Jun 15. Proteins. 2016. PMID: 27181425 Free PMC article.
-
Dockground: A comprehensive data resource for modeling of protein complexes.Protein Sci. 2018 Jan;27(1):172-181. doi: 10.1002/pro.3295. Epub 2017 Oct 10. Protein Sci. 2018. PMID: 28891124 Free PMC article.
References
-
- Aloy P, Pichaud M, Russell RB. Protein complexes: Structure prediction challenges for the 21st century. Curr Opin Struct Biol. 2005;15:15–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
