

A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function
- PMID: 25693964
- PMCID: PMC4403044
- DOI: 10.15252/emmm.201404576
A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function
Abstract
Loss of function of the FMR1 gene leads to fragile X syndrome (FXS), the most common form of intellectual disability. The loss of FMR1 function is usually caused by epigenetic silencing of the FMR1 promoter leading to expansion and subsequent methylation of a CGG repeat in the 5' untranslated region. Very few coding sequence variations have been experimentally characterized and shown to be causal to the disease. Here, we describe a novel FMR1 mutation and reveal an unexpected nuclear export function for the C-terminus of FMRP. We screened a cohort of patients with typical FXS symptoms who tested negative for CGG repeat expansion in the FMR1 locus. In one patient, we identified a guanine insertion in FMR1 exon 15. This mutation alters the open reading frame creating a short novel C-terminal sequence, followed by a stop codon. We find that this novel peptide encodes a functional nuclear localization signal (NLS) targeting the patient FMRP to the nucleolus in human cells. We also reveal an evolutionarily conserved nuclear export function associated with the endogenous C-terminus of FMRP. In vivo analyses in Drosophila demonstrate that a patient-mimetic mutation alters the localization and function of Dfmrp in neurons, leading to neomorphic neuronal phenotypes.
Keywords: Drosophila; axon guidance; fragile X syndrome; nuclear export; nucleolus.
© 2015 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
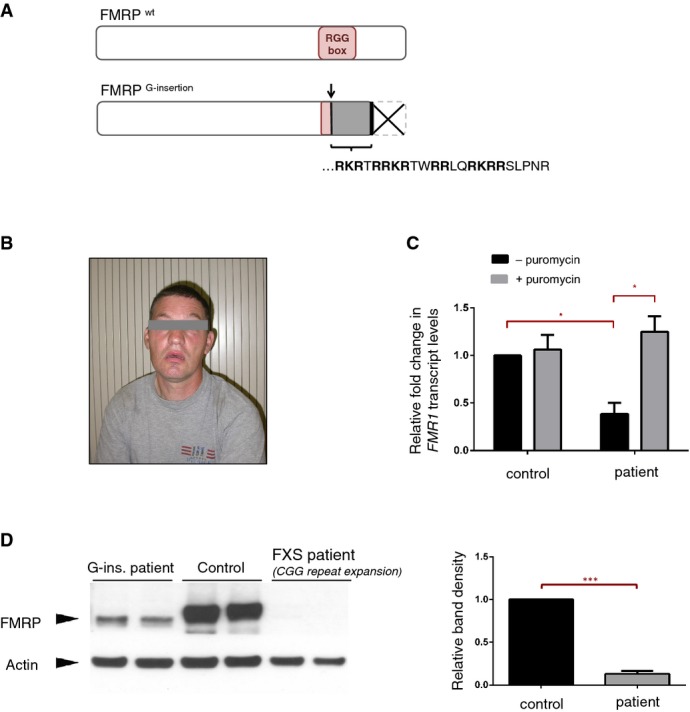
Predicted effects of the G-insertion mutation on FMRP are schematized. The insertion site corresponds to the beginning of the RNA-binding RGG Box, where it alters the open reading frame and thus disrupts the domain entirely. The frameshift creates a novel peptide sequence [RKRTRRKRTWRRLQRKRRSLPNR] followed by a premature stop codon, causing the truncation of the FMRP C-terminus.
Photograph of male patient with FMR1G-ins. allele. Typical physical and behavioral features of FXS were noted in the patient, who shows moderate to severe intellectual disability.
FMR1 mRNA levels were analyzed via RT–qPCR in EBV-transformed lymphocyte cells derived from the patient's blood samples. Patient cells showed a ˜60% decrease in FMR1 mRNA compared to control cells (*P = 0.010, 0.383 ± 0.110 SD, n = 3). Treatment with translational blocker puromycin restored FMR1 mRNA levels in patient cells (*P = 0.048, 0.383 ± 0.110 SD versus 1.25 ± 0.166 SD, n = 3), suggesting that the reduction of FMR1 mRNA in these cells is primarily due to nonsense-mediated decay. RT-qPCR reactions were run in triplicate in three independent experiments. Fold changes in FMR1 expression, normalized to HRPT expression, were calculated using the ΔΔCT method, and analyzed statistically with a two-tailed t-test (GraphPad). Error bars represent mean values with SD.
Western blot analysis shows a significant decrease (> 90%) in FMRP protein levels in patient-derived cells (***P = 0.008, 0.13 ± 0.031 SD, n = 2), and the patient FMRP migrates slightly lower than the wild-type protein. Band intensities for the different cell lines were quantified, normalized for actin and analyzed statistically with a twot-tailed t-test (GraphPad). Error bars represent mean values with SD.
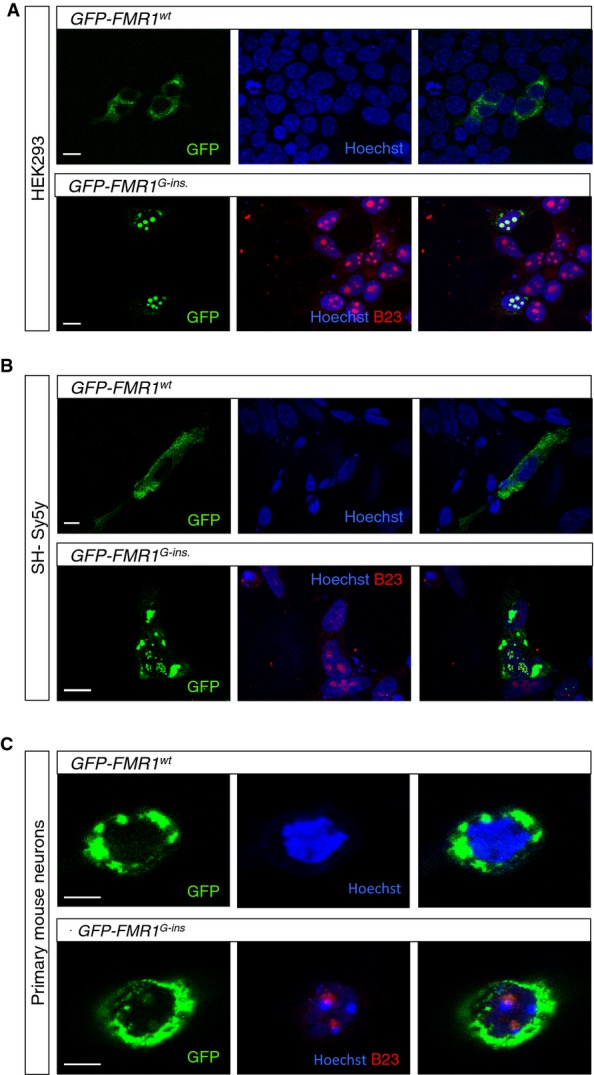
A–C HEK293 (A) and SH-Sy5y (B) cells, as well as primary neurons from FMR1 KO mice (C), were transfected with vectors encoding the GFP-tagged wild-type FMRP (GFP-FMR1WT) or the GFP-tagged patient FMRP (GFP-FMR1G-insertion) under the control of a β-actin promoter. Wild-type FMRP localizes predominantly in the cytoplasm, whereas the patient FMRP forms nuclear and nucleolar aggregates that colocalize with nucleophosmin. Scale bars in (A, B) represent 10 μm. Scale bar in (C) represents 5 μm.
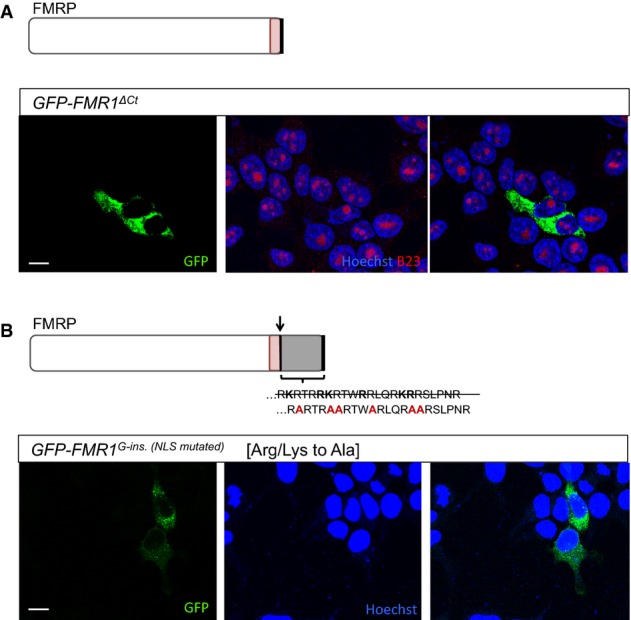
Transfected HEK293 cells expressing a GFP-tagged FMRP in which the C-terminus is truncated following the G-ins. mutation site (GFP-FMR1ΔCt). GFP-FMR1ΔCt protein is exclusively cytoplasmic, suggesting that the truncation of the C-terminus alone is not sufficient to explain the localization change observed for the patient FMRP.
Transfected HEK293 cells expressing a modified version of GFP-tagged patient FMRP (GFP-FMR1G-ins. [NLSmutated]). The novel amino acid sequence in the C-terminus of patient FMRP [RKRTRRKRTWRRLQRKRRSLPNR] contains stretches of arginines and lysines predicted to function as a nuclear localization signal (based on Expasy protein motif scanner, ScanProsite). Mutating adjacent R and K residues into alanines [RARTRAARTWARLQRAARSLPNR] abolishes the nucleolar localization of the patient FMRP, strongly suggesting that the novel amino acid sequence present in the patient FMRP C-terminus is a functional nuclear localization signal (NLS).
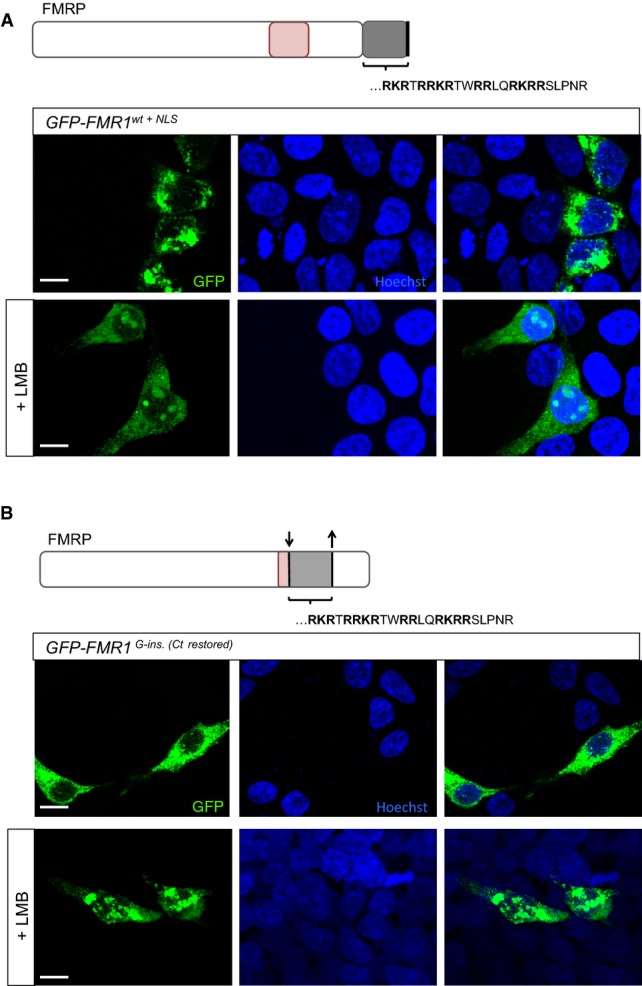
Transfected HEK293 cells expressing GFP-tagged wild-type FMRP fused to the patient NLS motif (GFP-FMR1wt+NLS). Unlike the patient FMRP, GFP-FMR1wt+NLS protein is predominantly cytoplasmic and does not aggregate in the nucleus. However, treatment of HEK293 cells expressing GFP-FMR1wt+NLS with leptomycin B—an inhibitor of nuclear export—resulted in the appearance of nucleolar inclusions. This suggests that the presence of a full-length C-terminus facilitates the nuclear export of FMRP in this context.
Transfected HEK293 cells expressing a modified version of GFP-tagged patient FMRP, in which the truncation of the C-terminus is reverted by restoring the open reading frame (GFP-FMR1G-ins.[Ct restored]). The patient protein is not detected in the nucleus when the C-terminus is intact; however, nucleolar retention of the protein is observed upon leptomycin B treatment of the transfected cells. In line with results from (A), this suggests that an intact C-terminus enables nuclear export of the FMR1 protein.
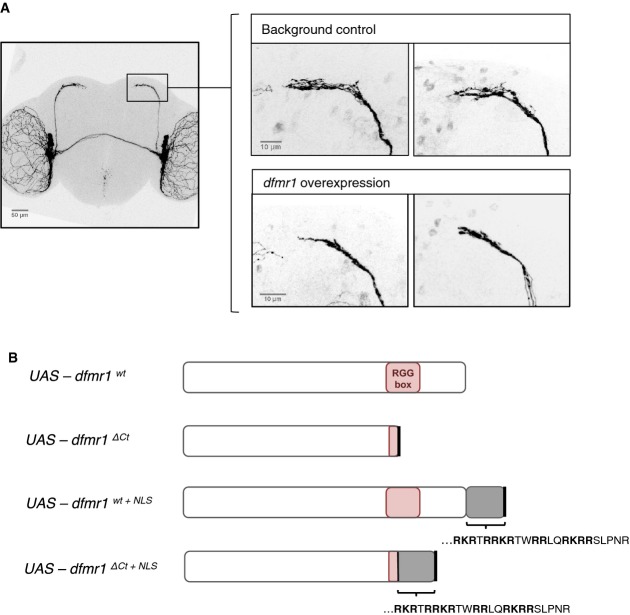
The morphology of small lateral ventral neurons (sLNvs) is sensitive to dfmr1 activity. Overexpression of wild-type dfmr1 in the sLNv neurons results in a consistent “axonal collapse” phenotype, where the branching of axonal termini of sLNvs is reduced. Scale bars represent 50 and 10 μm (magnified images).
Transgenic fly lines were created bearing UAS-dfmr1 variants, in order to assay in vivo functional changes associated with the effects of the patient mutation: the truncation of the C-terminus (dfmr1ΔCt), presence of the patient NLS sequence (dfmr1wt+NLS), or both (dfmr1ΔCt+NLS).
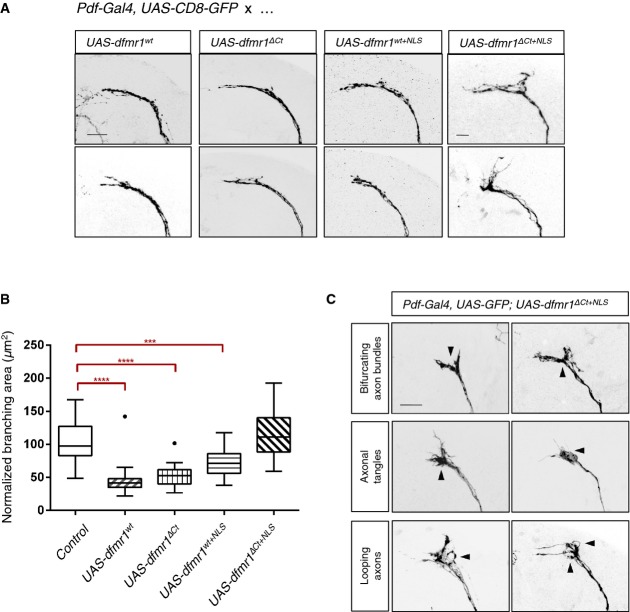
Overexpression of wild-type dfmr1 (UAS-dfmr1wt) using a LNv neuron-specific driver line (Pdf-Gal4) causes reduction of branching area—“axonal collapse”—in the sLNv axonal termini. dfmr1ΔCt and dfmr1wt+NLS overexpression phenocopies overexpression of dfmr1wt, suggesting that the truncation of the C-terminus or the presence of the patient NLS alone does not impair or alter the functional efficacy of dfmr1 in this context. However, overexpression of dfmr1ΔCt+NLS fails to cause collapse of sLNv termini, indicating a significant loss or change of Dfmrp protein function. Axonal morphology is visualized by expressing CD8-GFP under the control of the same neuronal driver. Scale bar represents 10 μm.
Quantification of axonal termini branching in LNv neurons overexpressing various dfmr1 alleles. Overexpression of dfmr1wt, dfmr1ΔCt, or dfmr1wt+NLS significantly reduced axonal branching area [****P < 0.0001; control 101.3 ± 29.76 SD, n = 30; UAS-dfmrwt 45.41 ± 21.69 SD, n = 29; UAS-dfmrΔCt 52.31 ± 14.67 SD, n = 32; UAS-dfmrwt+NLS 72.45 ± 21.83 SD, n = 29; UAS-dfmrΔCt+NLS 117.8 ± 36.96 SD, n = 28]. dfmr1ΔCt+NLS overexpression does not result in a significant change of axonal branching area compared to the background control [Pdf-Gal4, UAS-GFP] (P = 0.672). For quantification purposes, each branching area (n) was manually outlined, starting from the first point of defasculation of the axonal bundle. These measurements were normalized for variability in brain size across samples using LNv commissure length measurements for each brain. Two-tailed t-tests using Welch's correction were then used to compare controls with mutant phenotypes (GraphPad). Error bars represent mean values with SD. Dots visible for box plots of UAS-dfmrwt and UAS-dfmrΔCt represent samples that outlier the whiskers.
Overexpression of dfmr1ΔCt+NLS results in aberrant axonal termini morphology. Axonal guidance defects were frequently observed (arrowheads) and scored manually for bifurcations of the axon bundle (9/48), tangling of axons in the termini (13/48), and apparent looping of axons (7/48).
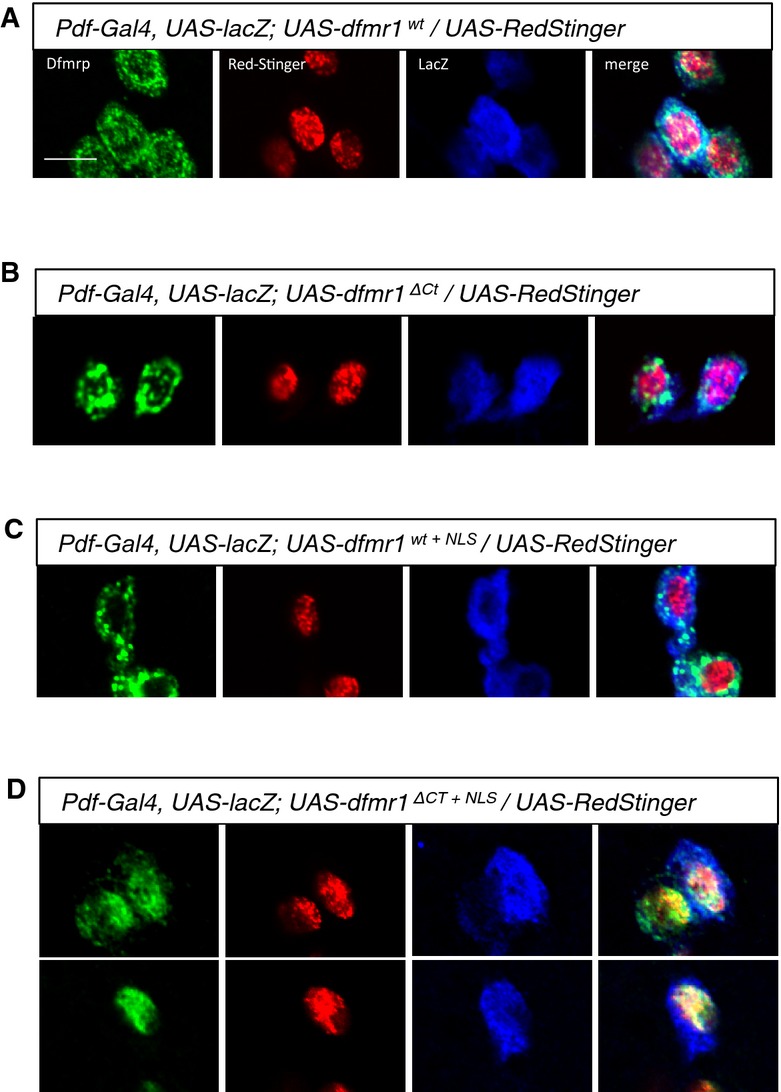
A–D Subcellular localization of transgenically expressed dfmr1wt, dfmr1ΔCt, dfmr1wt+NLS, and dfmr1ΔCt+NLS proteins in LNv neurons. DfmrpΔCt+NLS shows nuclear localization (A), colocalizing with the genetically encoded nuclear marker RedStinger, while Dfmrpwt, DfmrpΔCt, and Dfmrpwt+NLS are predominantly cytoplasmic (B–D). These results suggest that an intact C-terminus can mediate the nuclear export of the Dfmrp protein. Scale bar represents 5 μm.
Similar articles
-
CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons.Mol Autism. 2016 Oct 6;7:42. doi: 10.1186/s13229-016-0105-9. eCollection 2016. Mol Autism. 2016. PMID: 27713816 Free PMC article.
-
Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome.PLoS One. 2011;6(10):e26203. doi: 10.1371/journal.pone.0026203. Epub 2011 Oct 12. PLoS One. 2011. PMID: 22022567 Free PMC article.
-
Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures.Proc Natl Acad Sci U S A. 2015 Jan 27;112(4):949-56. doi: 10.1073/pnas.1423094112. Epub 2015 Jan 5. Proc Natl Acad Sci U S A. 2015. PMID: 25561520 Free PMC article.
-
Human pluripotent stem cell models of Fragile X syndrome.Mol Cell Neurosci. 2016 Jun;73:43-51. doi: 10.1016/j.mcn.2015.11.011. Epub 2015 Nov 27. Mol Cell Neurosci. 2016. PMID: 26640241 Free PMC article. Review.
-
[Chromatin changes caused by CGG repeat expansion in fmr1 gene].Mol Biol (Mosk). 2015 Mar-Apr;49(2):205-11. Mol Biol (Mosk). 2015. PMID: 26065250 Review. Russian.
Cited by
-
Loss of ZNF32 augments the regeneration of nervous lateral line system through negative regulation of SOX2 transcription.Oncotarget. 2016 Oct 25;7(43):70420-70436. doi: 10.18632/oncotarget.11895. Oncotarget. 2016. PMID: 27626680 Free PMC article.
-
Function of FMRP Domains in Regulating Distinct Roles of Neuronal Protein Synthesis.Mol Neurobiol. 2022 Dec;59(12):7370-7392. doi: 10.1007/s12035-022-03049-1. Epub 2022 Oct 1. Mol Neurobiol. 2022. PMID: 36181660
-
Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes.Genes (Basel). 2021 Oct 22;12(11):1669. doi: 10.3390/genes12111669. Genes (Basel). 2021. PMID: 34828275 Free PMC article.
-
Drosophila melanogaster as a Model to Study the Multiple Phenotypes, Related to Genome Stability of the Fragile-X Syndrome.Front Genet. 2019 Feb 13;10:10. doi: 10.3389/fgene.2019.00010. eCollection 2019. Front Genet. 2019. PMID: 30815010 Free PMC article. Review.
-
Mouse models of fragile X-related disorders.Dis Model Mech. 2023 Feb 1;16(2):dmm049485. doi: 10.1242/dmm.049485. Epub 2023 Jan 24. Dis Model Mech. 2023. PMID: 36692473 Free PMC article. Review.
References
-
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. - PubMed
-
- Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258:162–170. - PubMed
-
- Belser RC, Sudhalter V. Conversational characteristics of children with fragile X syndrome: repetitive speech. Am J Ment Retard. 2001;106:28–38. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
