

Template-based prediction of protein function
- PMID: 25678152
- PMCID: PMC4512936
- DOI: 10.1016/j.sbi.2015.01.007
Template-based prediction of protein function
Abstract
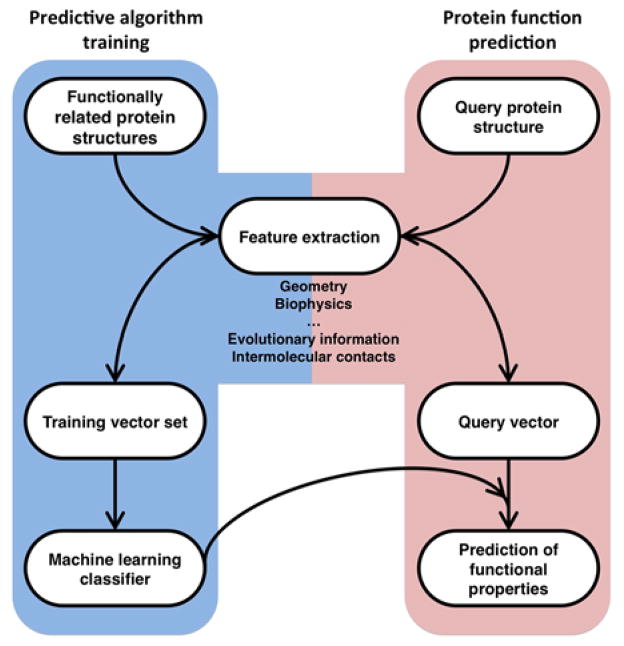
We discuss recent approaches for structure-based protein function annotation. We focus on template-based methods where the function of a query protein is deduced from that of a template for which both the structure and function are known. We describe the different ways of identifying a template. These are typically based on sequence analysis but new methods based on purely structural similarity are also being developed that allow function annotation based on structural relationships that cannot be recognized by sequence. The growing number of available structures of known function, improved homology modeling techniques and new developments in the use of structure allow template-based methods to be applied on a proteome-wide scale and in many different biological contexts. This progress significantly expands the range of applicability of structural information in function annotation to a level that previously was only achievable by sequence comparison.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures

Similar articles
-
Prediction of enzyme function based on 3D templates of evolutionarily important amino acids.BMC Bioinformatics. 2008 Jan 11;9:17. doi: 10.1186/1471-2105-9-17. BMC Bioinformatics. 2008. PMID: 18190718 Free PMC article.
-
Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre.Proteins. 2008 Feb 15;70(3):611-25. doi: 10.1002/prot.21688. Proteins. 2008. PMID: 17876813
-
DNABind: a hybrid algorithm for structure-based prediction of DNA-binding residues by combining machine learning- and template-based approaches.Proteins. 2013 Nov;81(11):1885-99. doi: 10.1002/prot.24330. Epub 2013 Aug 16. Proteins. 2013. PMID: 23737141
-
In quest of an empirical potential for protein structure prediction.Curr Opin Struct Biol. 2006 Apr;16(2):166-71. doi: 10.1016/j.sbi.2006.02.004. Epub 2006 Mar 9. Curr Opin Struct Biol. 2006. PMID: 16524716 Review.
-
Protein structure prediction in structure-based ligand design and virtual screening.Comb Chem High Throughput Screen. 2009 Dec;12(10):940-60. doi: 10.2174/138620709789824718. Comb Chem High Throughput Screen. 2009. PMID: 20025561 Review.
Cited by
-
Functional prediction of hypothetical proteins in human adenoviruses.Bioinformation. 2015 Oct 31;11(10):466-73. doi: 10.6026/97320630011466. eCollection 2015. Bioinformation. 2015. PMID: 26664031 Free PMC article.
-
Modeling enzyme-ligand binding in drug discovery.J Cheminform. 2015 Oct 6;7(1):48. doi: 10.1186/s13321-015-0096-0. eCollection 2015 Dec. J Cheminform. 2015. PMID: 26457119 Free PMC article. Review.
-
Modeling complexes of modeled proteins.Proteins. 2017 Mar;85(3):470-478. doi: 10.1002/prot.25183. Epub 2016 Oct 24. Proteins. 2017. PMID: 27701777 Free PMC article.
-
Functional classification of protein structures by local structure matching in graph representation.Protein Sci. 2018 Jun;27(6):1125-1135. doi: 10.1002/pro.3416. Epub 2018 Apr 27. Protein Sci. 2018. PMID: 29604149 Free PMC article.
-
Cell cycle gene expression networks discovered using systems biology: Significance in carcinogenesis.J Cell Physiol. 2015 Oct;230(10):2533-42. doi: 10.1002/jcp.24990. J Cell Physiol. 2015. PMID: 25808367 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
