

PCalign: a method to quantify physicochemical similarity of protein-protein interfaces
- PMID: 25638036
- PMCID: PMC4339745
- DOI: 10.1186/s12859-015-0471-x
PCalign: a method to quantify physicochemical similarity of protein-protein interfaces
Abstract
Background: Structural comparison of protein-protein interfaces provides valuable insights into the functional relationship between proteins, which may not solely arise from shared evolutionary origin. A few methods that exist for such comparative studies have focused on structural models determined at atomic resolution, and may miss out interesting patterns present in large macromolecular complexes that are typically solved by low-resolution techniques.
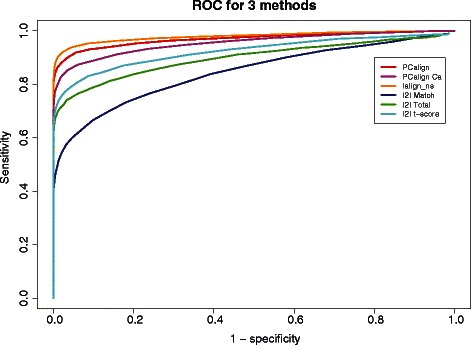
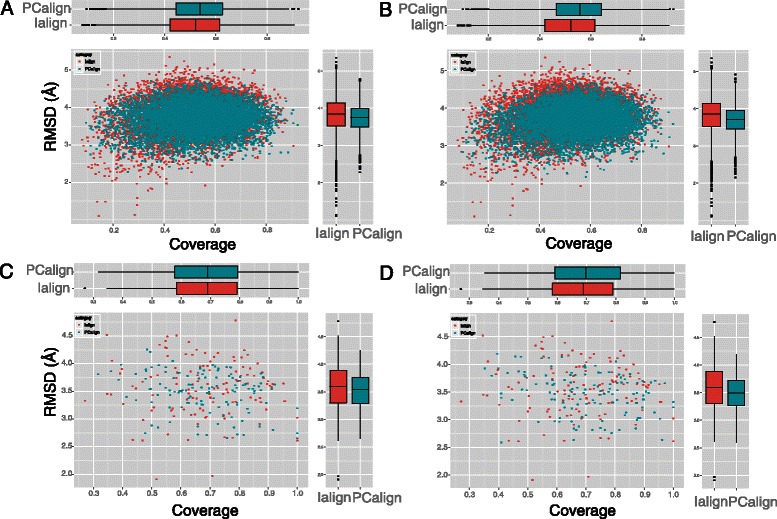
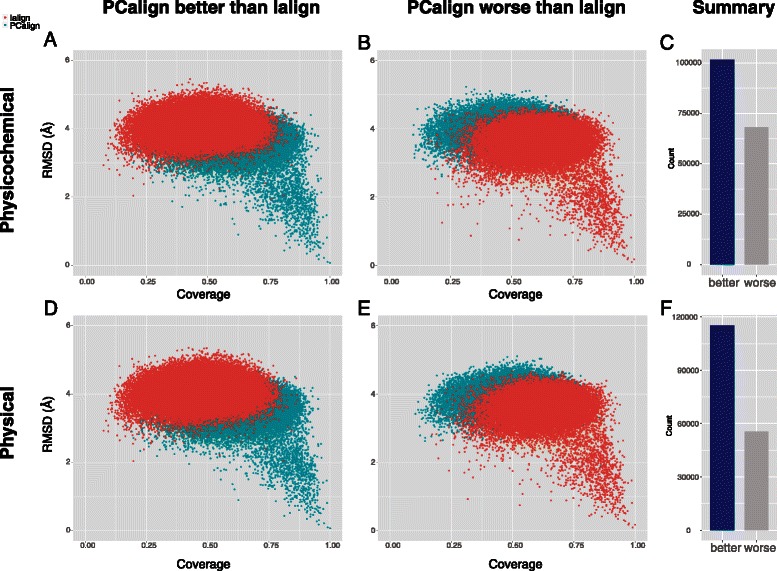
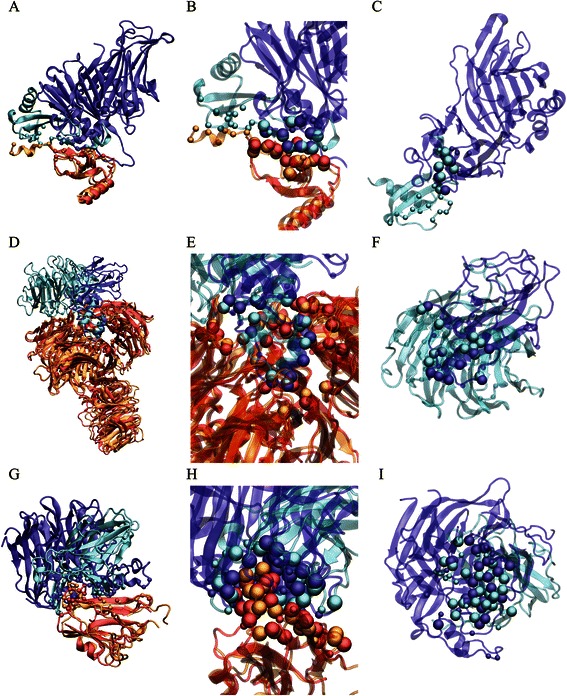
Results: We developed a coarse-grained method, PCalign, to quantitatively evaluate physicochemical similarities between a given pair of protein-protein interfaces. This method uses an order-independent algorithm, geometric hashing, to superimpose the backbone atoms of a given pair of interfaces, and provides a normalized scoring function, PC-score, to account for the extent of overlap in terms of both geometric and chemical characteristics. We demonstrate that PCalign outperforms existing methods, and additionally facilitates comparative studies across models of different resolutions, which are not accommodated by existing methods. Furthermore, we illustrate potential application of our method to recognize interesting biological relationships masked by apparent lack of structural similarity.
Conclusions: PCalign is a useful method in recognizing shared chemical and spatial patterns among protein-protein interfaces. It outperforms existing methods for high-quality data, and additionally facilitates comparison across structural models with different levels of details with proven robustness against noise.
Figures
Similar articles
-
Protein-Protein Interfaces in Viral Capsids Are Structurally Unique.J Mol Biol. 2015 Nov 6;427(22):3613-3624. doi: 10.1016/j.jmb.2015.09.008. Epub 2015 Sep 12. J Mol Biol. 2015. PMID: 26375252 Free PMC article.
-
Structural motifs at protein-protein interfaces: protein cores versus two-state and three-state model complexes.Protein Sci. 1997 Sep;6(9):1793-805. doi: 10.1002/pro.5560060901. Protein Sci. 1997. PMID: 9300480 Free PMC article. Review.
-
Structural alignment of protein--DNA interfaces: insights into the determinants of binding specificity.J Mol Biol. 2005 Feb 4;345(5):1027-45. doi: 10.1016/j.jmb.2004.11.010. Epub 2004 Dec 8. J Mol Biol. 2005. PMID: 15644202
-
Generation and analysis of a protein-protein interface data set with similar chemical and spatial patterns of interactions.Proteins. 2005 Oct 1;61(1):6-20. doi: 10.1002/prot.20580. Proteins. 2005. PMID: 16184518
-
PocketAlign a novel algorithm for aligning binding sites in protein structures.J Chem Inf Model. 2011 Jul 25;51(7):1725-36. doi: 10.1021/ci200132z. Epub 2011 Jun 21. J Chem Inf Model. 2011. PMID: 21662242
Cited by
-
Predicting Protein-Peptide Complex Structures by Accounting for Peptide Flexibility and the Physicochemical Environment.J Chem Inf Model. 2022 Jan 10;62(1):27-39. doi: 10.1021/acs.jcim.1c00836. Epub 2021 Dec 21. J Chem Inf Model. 2022. PMID: 34931833 Free PMC article.
-
Protein-Protein Interfaces in Viral Capsids Are Structurally Unique.J Mol Biol. 2015 Nov 6;427(22):3613-3624. doi: 10.1016/j.jmb.2015.09.008. Epub 2015 Sep 12. J Mol Biol. 2015. PMID: 26375252 Free PMC article.
-
Capturing a Dynamic Chaperone-Substrate Interaction Using NMR-Informed Molecular Modeling.J Am Chem Soc. 2016 Aug 10;138(31):9826-39. doi: 10.1021/jacs.6b02382. Epub 2016 Aug 2. J Am Chem Soc. 2016. PMID: 27415450 Free PMC article.
-
G-LoSA: An efficient computational tool for local structure-centric biological studies and drug design.Protein Sci. 2016 Apr;25(4):865-76. doi: 10.1002/pro.2890. Epub 2016 Mar 6. Protein Sci. 2016. PMID: 26813336 Free PMC article.
-
PatchSearch: a web server for off-target protein identification.Nucleic Acids Res. 2019 Jul 2;47(W1):W365-W372. doi: 10.1093/nar/gkz478. Nucleic Acids Res. 2019. PMID: 31131411 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
