

Molecular mechanisms of autophagy in the cardiovascular system
- PMID: 25634969
- PMCID: PMC4313620
- DOI: 10.1161/CIRCRESAHA.114.303788
Molecular mechanisms of autophagy in the cardiovascular system
Erratum in
-
Correction.Circ Res. 2015 Mar 27;116(7):e56. doi: 10.1161/RES.0000000000000052. Circ Res. 2015. PMID: 25814691 No abstract available.
Abstract
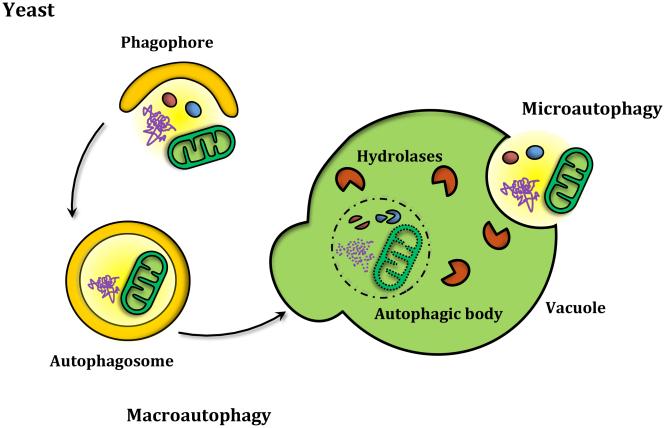
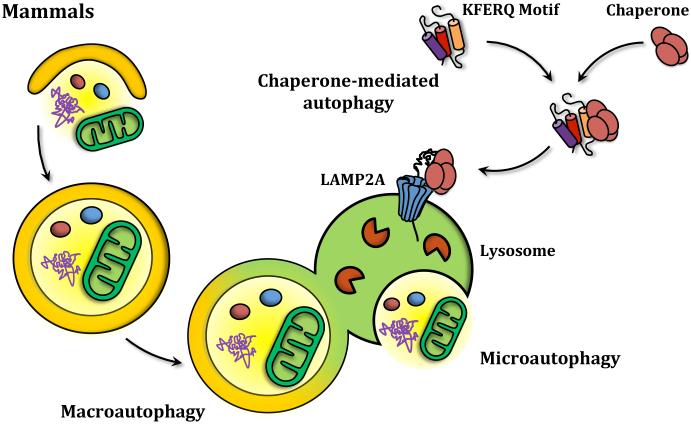
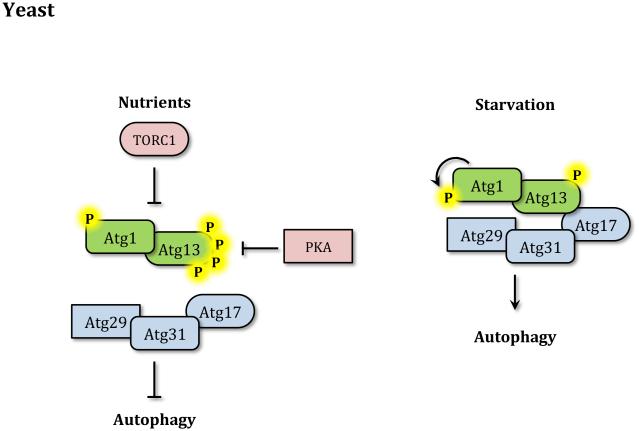
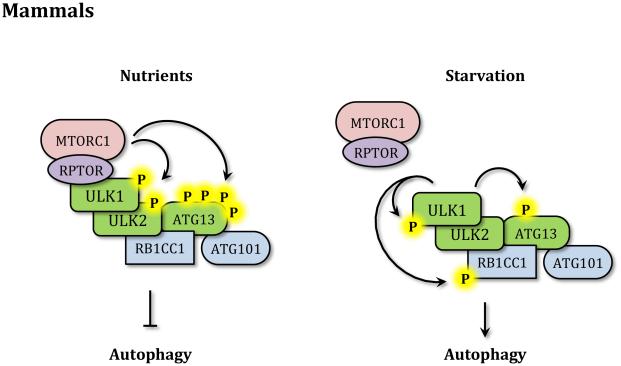
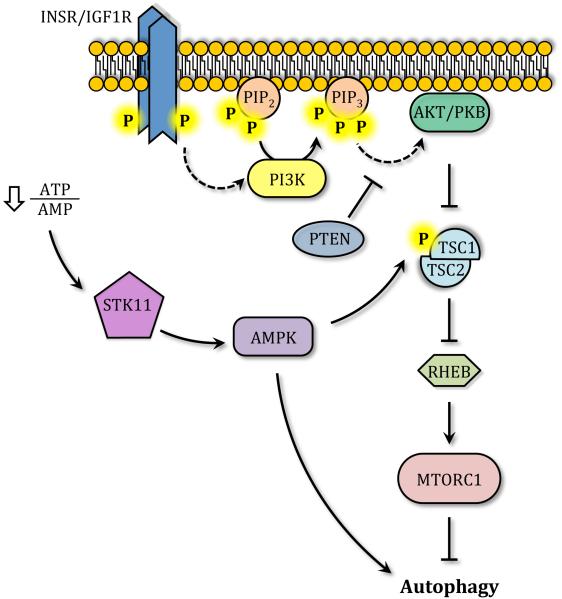
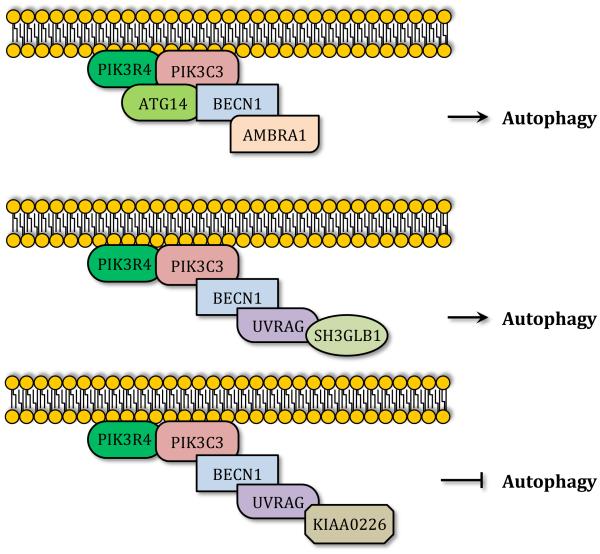
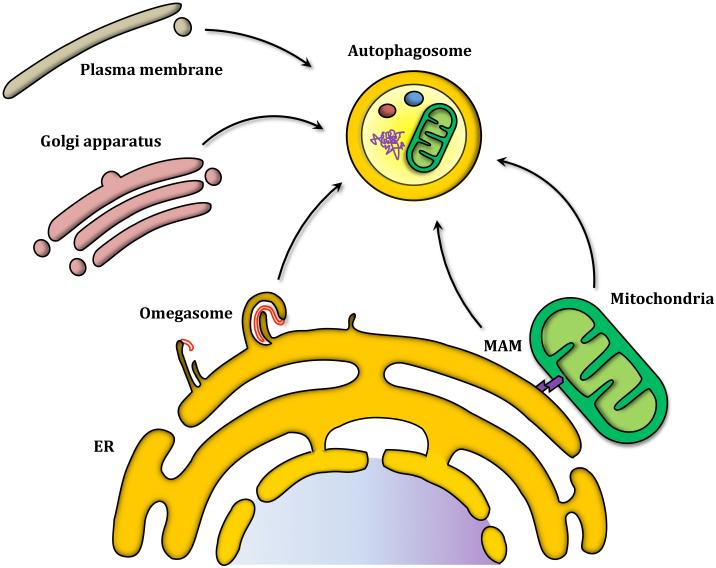
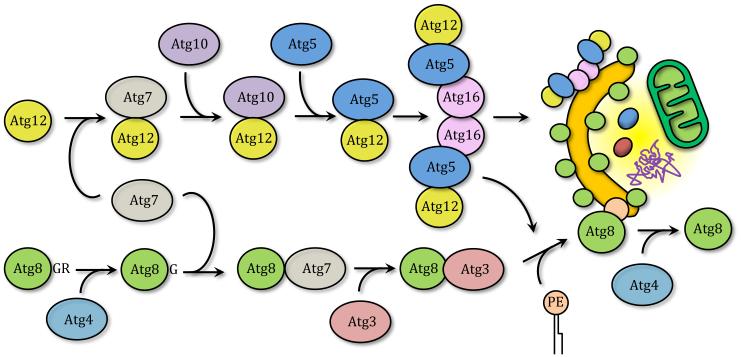
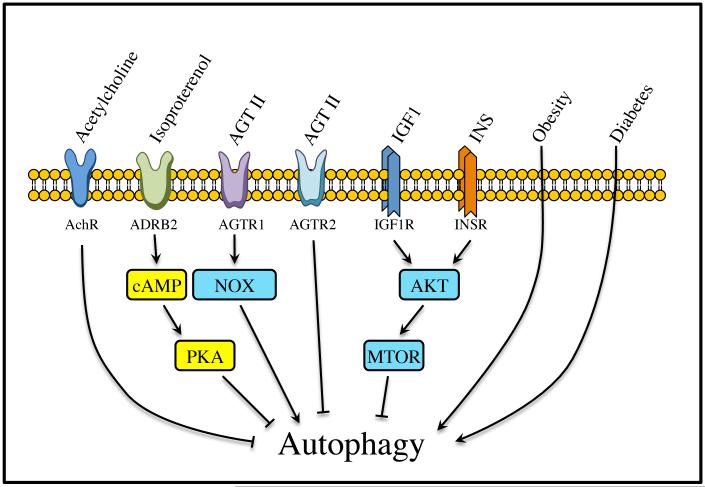
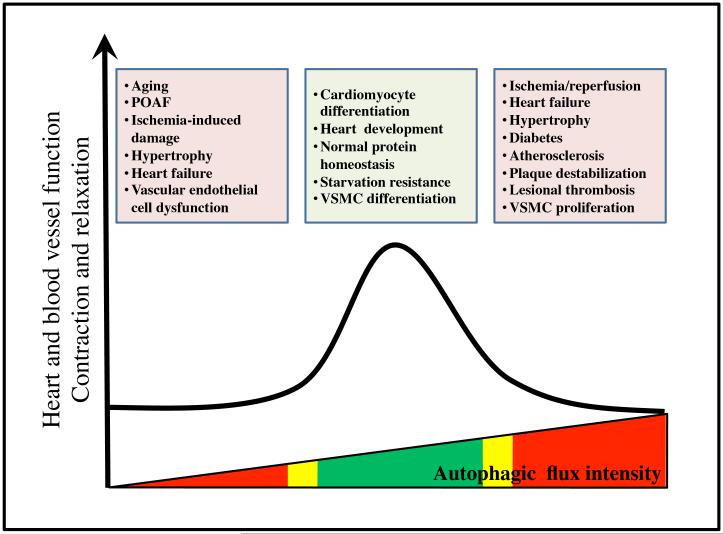
Autophagy is a catabolic recycling pathway triggered by various intra- or extracellular stimuli that is conserved from yeast to mammals. During autophagy, diverse cytosolic constituents are enveloped by double-membrane vesicles, autophagosomes, which later fuse with lysosomes or the vacuole to degrade their cargo. Dysregulation in autophagy is associated with a diverse range of pathologies including cardiovascular disease, the leading cause of death in the world. As such, there is great interest in identifying novel mechanisms that govern the cardiovascular response to disease-related stress. First described in failing hearts, autophagy within the cardiovascular system has been characterized widely in cardiomyocytes, cardiac fibroblasts, endothelial cells, and vascular smooth muscle cells. In all cases, a window of optimal autophagic activity seems to be critical to the maintenance of cardiovascular homeostasis and function; excessive or insufficient levels of autophagic flux can each contribute to heart disease pathogenesis. Here, we review the molecular mechanisms that govern autophagosome formation and analyze the link between autophagy and cardiovascular disease.
Keywords: atherosclerosis; blood vessels; cardiovascular diseases; heart; intracellular signaling proteins; muscle, smooth, vascular; myocytes, cardiac.
© 2015 American Heart Association, Inc.
Figures
Similar articles
-
Autophagy in cardiovascular biology.J Clin Invest. 2015 Jan;125(1):55-64. doi: 10.1172/JCI73943. Epub 2015 Jan 2. J Clin Invest. 2015. PMID: 25654551 Free PMC article. Review.
-
Therapeutic targeting of autophagy in cardiovascular disease.J Mol Cell Cardiol. 2016 Jun;95:86-93. doi: 10.1016/j.yjmcc.2015.11.019. Epub 2015 Nov 18. J Mol Cell Cardiol. 2016. PMID: 26602750 Free PMC article. Review.
-
Cardiovascular autophagy: concepts, controversies, and perspectives.Autophagy. 2013 Oct;9(10):1455-66. doi: 10.4161/auto.25969. Epub 2013 Aug 14. Autophagy. 2013. PMID: 23959233 Review.
-
Autophagy in cardiac metabolic control: Novel mechanisms for cardiovascular disorders.Cell Biol Int. 2016 Sep;40(9):944-54. doi: 10.1002/cbin.10626. Epub 2016 Jul 4. Cell Biol Int. 2016. PMID: 27191043 Review.
-
The role of autophagy in cardiovascular pathology.Cardiovasc Res. 2022 Mar 16;118(4):934-950. doi: 10.1093/cvr/cvab158. Cardiovasc Res. 2022. PMID: 33956077 Free PMC article. Review.
Cited by
-
Autosis: A New Target to Prevent Cell Death.JACC Basic Transl Sci. 2020 Aug 24;5(8):857-869. doi: 10.1016/j.jacbts.2020.04.014. eCollection 2020 Aug. JACC Basic Transl Sci. 2020. PMID: 32875173 Free PMC article. Review.
-
Proteotoxicity and cardiac dysfunction.Circ Res. 2015 May 22;116(11):1863-82. doi: 10.1161/CIRCRESAHA.116.305372. Circ Res. 2015. PMID: 25999425 Free PMC article. Review.
-
Interplay between Exosomes and Autophagy in Cardiovascular Diseases: Novel Promising Target for Diagnostic and Therapeutic Application.Aging Dis. 2019 Dec 1;10(6):1302-1310. doi: 10.14336/AD.2018.1020. eCollection 2019 Dec. Aging Dis. 2019. PMID: 31788341 Free PMC article. Review.
-
The Impact of Moderate Chronic Hypoxia and Hyperoxia on the Level of Apoptotic and Autophagic Proteins in Myocardial Tissue.Oxid Med Cell Longev. 2018 Aug 16;2018:5786742. doi: 10.1155/2018/5786742. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 30186545 Free PMC article.
-
Paclitaxel alleviates monocrotaline-induced pulmonary arterial hypertension via inhibition of FoxO1-mediated autophagy.Naunyn Schmiedebergs Arch Pharmacol. 2019 May;392(5):605-613. doi: 10.1007/s00210-019-01615-4. Epub 2019 Jan 25. Naunyn Schmiedebergs Arch Pharmacol. 2019. PMID: 30683943
References
-
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–182. - PubMed
-
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting bcg and mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
