

Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome
- PMID: 25620203
- PMCID: PMC4320253
- DOI: 10.1016/j.ajhg.2014.11.019
Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome
Abstract
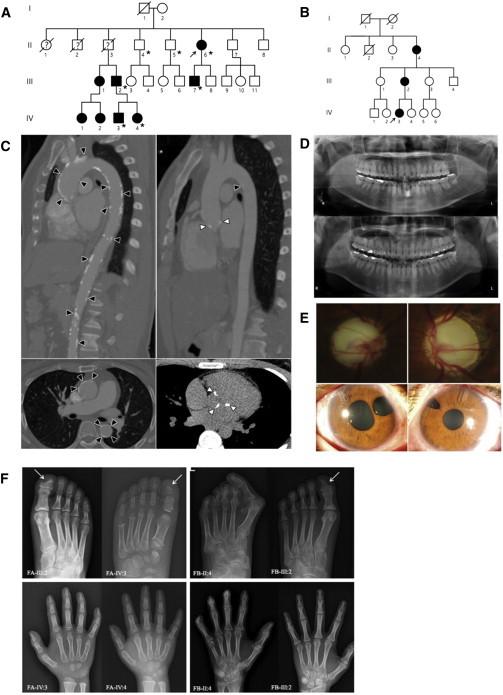
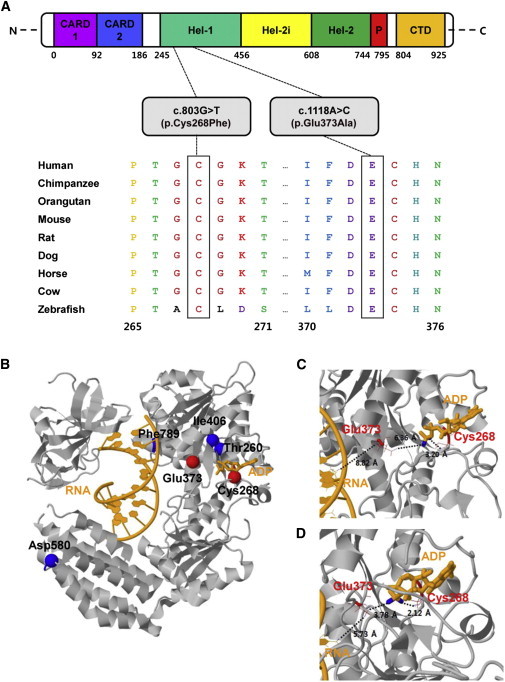
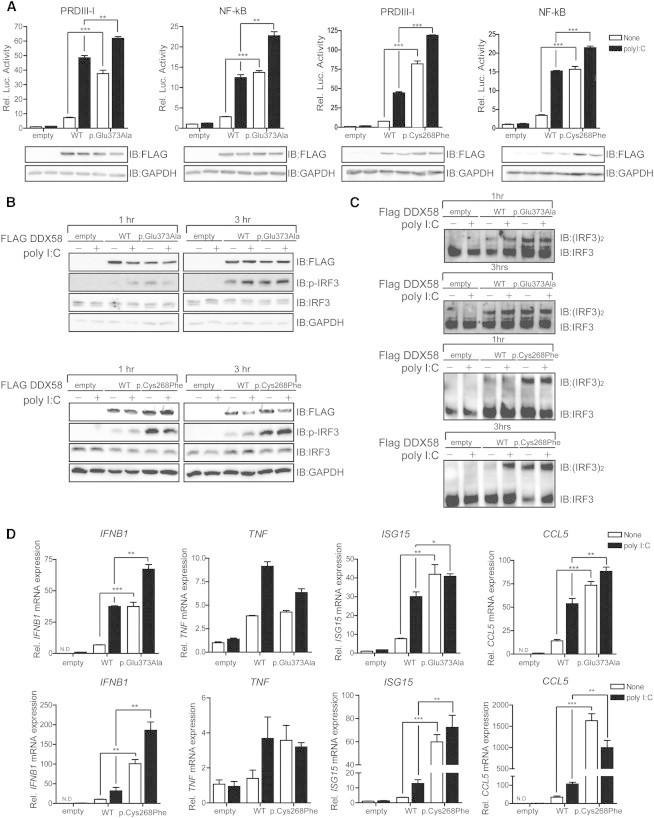
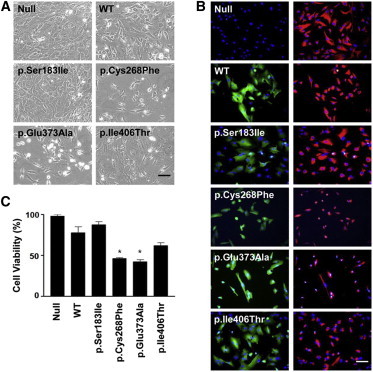
Singleton-Merten syndrome (SMS) is an autosomal-dominant multi-system disorder characterized by dental dysplasia, aortic calcification, skeletal abnormalities, glaucoma, psoriasis, and other conditions. Despite an apparent autosomal-dominant pattern of inheritance, the genetic background of SMS and information about its phenotypic heterogeneity remain unknown. Recently, we found a family affected by glaucoma, aortic calcification, and skeletal abnormalities. Unlike subjects with classic SMS, affected individuals showed normal dentition, suggesting atypical SMS. To identify genetic causes of the disease, we performed exome sequencing in this family and identified a variant (c.1118A>C [p.Glu373Ala]) of DDX58, whose protein product is also known as RIG-I. Further analysis of DDX58 in 100 individuals with congenital glaucoma identified another variant (c.803G>T [p.Cys268Phe]) in a family who harbored neither dental anomalies nor aortic calcification but who suffered from glaucoma and skeletal abnormalities. Cys268 and Glu373 residues of DDX58 belong to ATP-binding motifs I and II, respectively, and these residues are predicted to be located closer to the ADP and RNA molecules than other nonpathogenic missense variants by protein structure analysis. Functional assays revealed that DDX58 alterations confer constitutive activation and thus lead to increased interferon (IFN) activity and IFN-stimulated gene expression. In addition, when we transduced primary human trabecular meshwork cells with c.803G>T (p.Cys268Phe) and c.1118A>C (p.Glu373Ala) mutants, cytopathic effects and a significant decrease in cell number were observed. Taken together, our results demonstrate that DDX58 mutations cause atypical SMS manifesting with variable expression of glaucoma, aortic calcification, and skeletal abnormalities without dental anomalies.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome.Am J Hum Genet. 2015 Feb 5;96(2):275-82. doi: 10.1016/j.ajhg.2014.12.014. Epub 2015 Jan 22. Am J Hum Genet. 2015. PMID: 25620204 Free PMC article.
-
A novel IFIH1 mutation in the pincer domain underlies the clinical features of both Aicardi-Goutières and Singleton-Merten syndromes in a single patient.Br J Dermatol. 2018 Feb;178(2):e111-e113. doi: 10.1111/bjd.15869. Epub 2017 Dec 22. Br J Dermatol. 2018. PMID: 29270977 No abstract available.
-
DDX58 and Classic Singleton-Merten Syndrome.J Clin Immunol. 2019 Jan;39(1):75-80. doi: 10.1007/s10875-018-0572-1. Epub 2018 Dec 20. J Clin Immunol. 2019. PMID: 30574673 Free PMC article.
-
MDA5-Associated Neuroinflammation and the Singleton-Merten Syndrome: Two Faces of the Same Type I Interferonopathy Spectrum.J Interferon Cytokine Res. 2017 May;37(5):214-219. doi: 10.1089/jir.2017.0004. J Interferon Cytokine Res. 2017. PMID: 28475458 Free PMC article. Review.
-
RIG-I-Like Receptor Signaling in Singleton-Merten Syndrome.Front Genet. 2017 Sep 12;8:118. doi: 10.3389/fgene.2017.00118. eCollection 2017. Front Genet. 2017. PMID: 28955379 Free PMC article. Review.
Cited by
-
Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models.Proc Natl Acad Sci U S A. 2019 Apr 16;116(16):7941-7950. doi: 10.1073/pnas.1818281116. Epub 2019 Apr 3. Proc Natl Acad Sci U S A. 2019. PMID: 30944222 Free PMC article.
-
Type I interferon in rheumatic diseases.Nat Rev Rheumatol. 2018 Mar 21;14(4):214-228. doi: 10.1038/nrrheum.2018.31. Nat Rev Rheumatol. 2018. PMID: 29559718 Free PMC article. Review.
-
A GTPase-activating protein-binding protein (G3BP1)/antiviral protein relay conveys arteriosclerotic Wnt signals in aortic smooth muscle cells.J Biol Chem. 2018 May 25;293(21):7942-7968. doi: 10.1074/jbc.RA118.002046. Epub 2018 Apr 6. J Biol Chem. 2018. PMID: 29626090 Free PMC article.
-
Nucleic Acid Sensing in the Tumor Vasculature.Cancers (Basel). 2021 Sep 3;13(17):4452. doi: 10.3390/cancers13174452. Cancers (Basel). 2021. PMID: 34503262 Free PMC article. Review.
-
Translating nucleic acid-sensing pathways into therapies.Nat Rev Immunol. 2015 Sep 15;15(9):529-44. doi: 10.1038/nri3875. Epub 2015 Aug 21. Nat Rev Immunol. 2015. PMID: 26292638 Review.
References
-
- Singleton E.B., Merten D.F. An unusual syndrome of widened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr. Radiol. 1973;1:2–7. - PubMed
-
- Feigenbaum A., Müller C., Yale C., Kleinheinz J., Jezewski P., Kehl H.G., MacDougall M., Rutsch F., Hennekam R.C. Singleton-Merten syndrome: an autosomal dominant disorder with variable expression. Am. J. Med. Genet. A. 2013;161A:360–370. - PubMed
-
- Gay B.B., Jr., Kuhn J.P. A syndrome of widened medullary cavities of bone, aortic calcification, abnormal dentition, and muscular weakness (the Singleton-Merten syndrome) Radiology. 1976;118:389–395. - PubMed
-
- Valverde I., Rosenthal E., Tzifa A., Desai P., Bell A., Pushparajah K., Qureshi S., Beerbaum P., Simpson J. Singleton-merten syndrome and impaired cardiac function. J. Am. Coll. Cardiol. 2010;56:1760. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
