

MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies
- PMID: 25590803
- PMCID: PMC4669737
- DOI: 10.1038/cddis.2014.525
MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies
Erratum in
-
Correction to: MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies.Cell Death Dis. 2024 Jul 24;15(7):526. doi: 10.1038/s41419-024-06878-1. Cell Death Dis. 2024. PMID: 39048569 Free PMC article. No abstract available.
Abstract
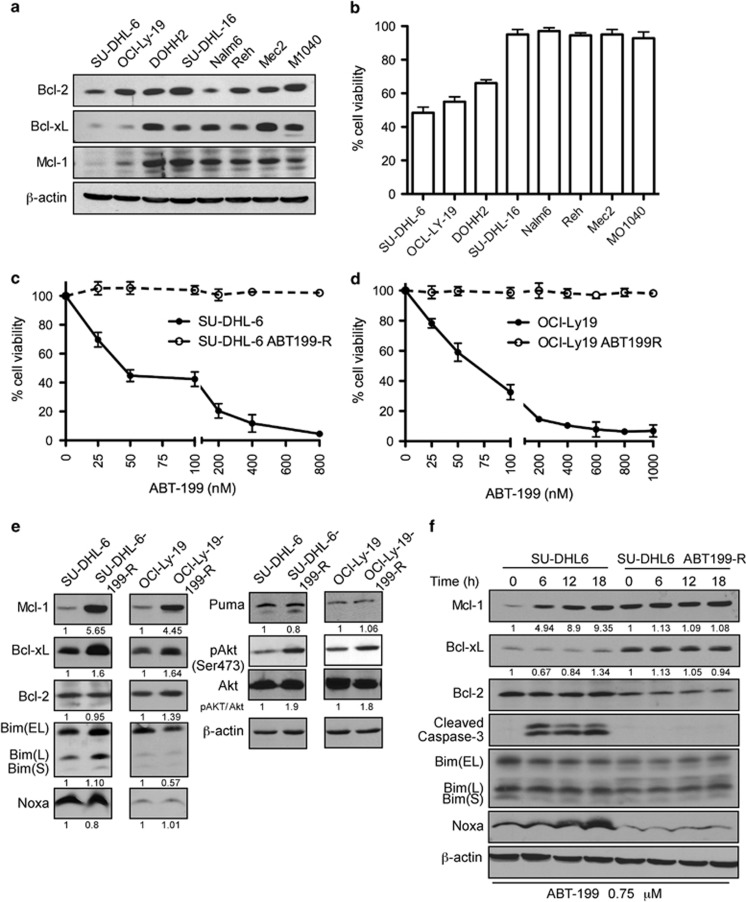
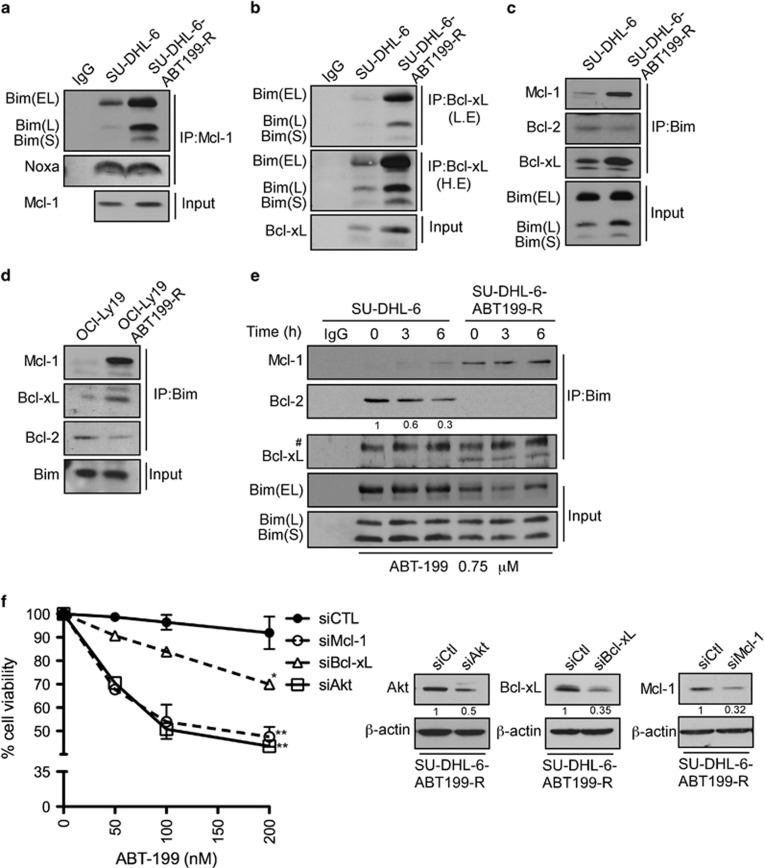
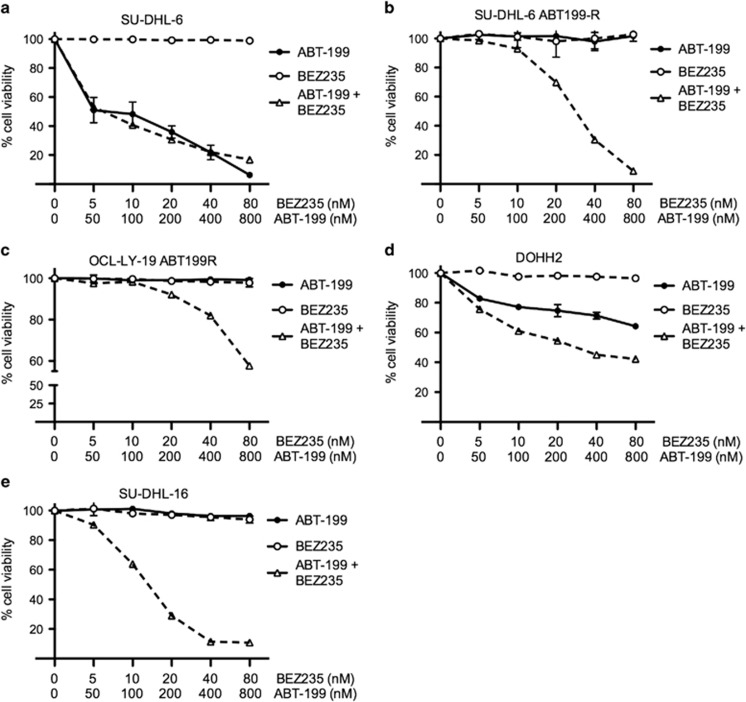
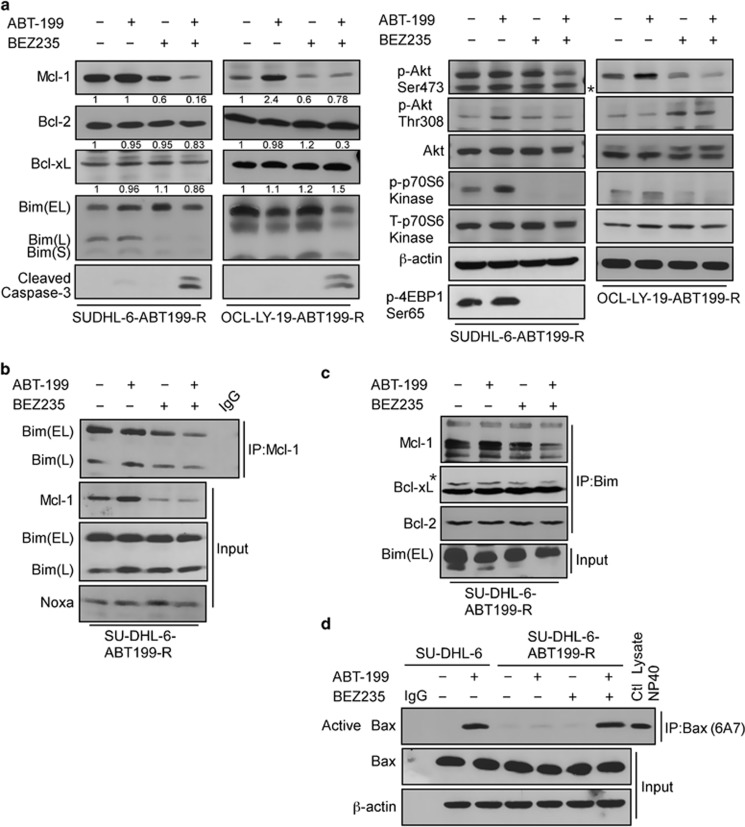
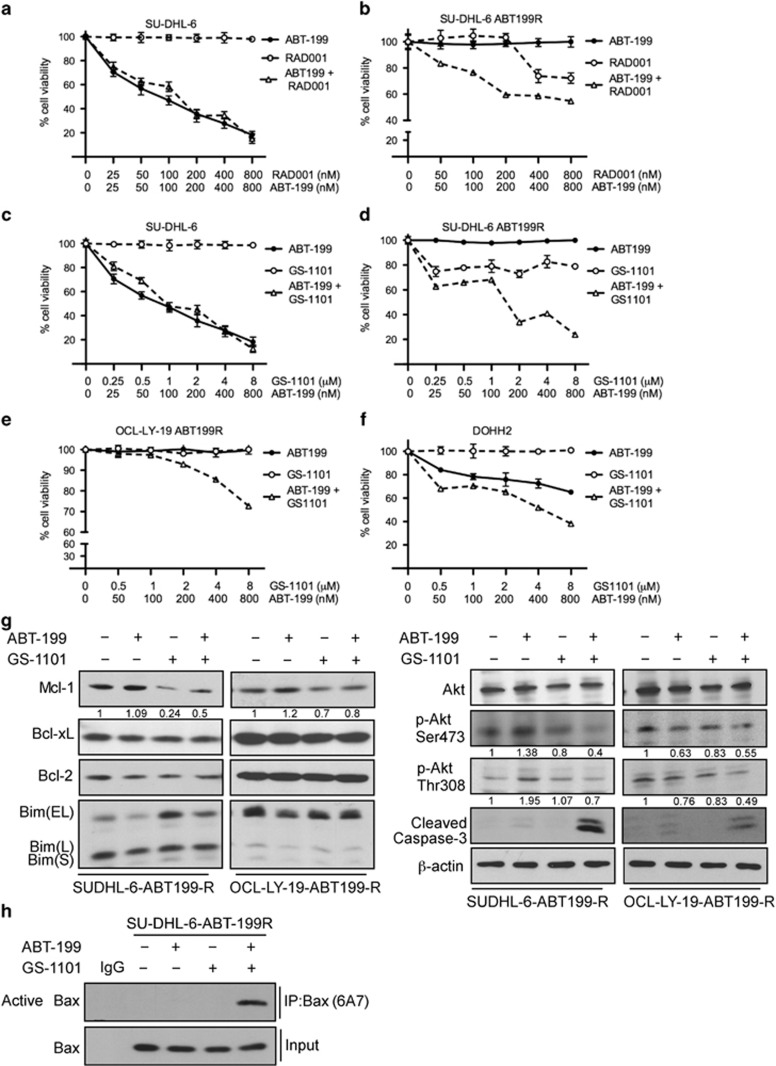
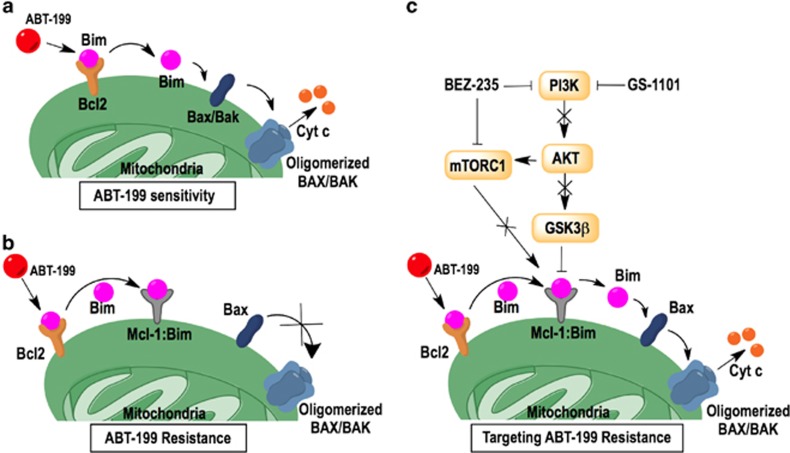
Overexpression of anti-apoptotic BCL-2 family members is a hallmark of many lymphoid malignancies, including chronic lymphocytic leukemia (CLL) and non-Hodgkin lymphoma (NHL) that can be targeted with small molecule inhibitors. ABT-199 is a rationally designed BCL-2 homology (BH)-3 mimetic that specifically binds to BCL-2, but not to MCL-1 and BCL-xL. Although the thrombocytopenia that occurs with navitoclax treatment has not been a problem with ABT-199, clinical trials in CLL could benefit by lowering the ABT-199 concentration through targeting other survival pathways. In this study, we investigated the mechanisms of resistance that develops to ABT-199 therapy by generating ABT-199-resistant (ABT199-R) cell lines via chronic exposure of NHL cell lines to ABT-199. Acquired resistance resulted in substantial AKT activation and upregulation of MCL-1 and BCL-xL levels that sequestered BIM. ABT199-R cells exhibited increased MCL-1 stability and failed to activate BAX in response to ABT-199. The ABT-199 acquired and inherent resistant cells were sensitized to treatment with ABT-199 by inhibitors of the PI3K, AKT, and mTOR pathways, NVP-BEZ235 and GS-1101. NVP-BEZ235, a dual inhibitor of p-AKT and mTOR, reduced MCL-1 levels causing BIM release from MCL-1 and BCL-xL, thus leading to cell death by BAX activation. The PI3Kδ inhibitor GS-1101 (idelalisib) downregulated MCL-1 and sensitized ABT199-R cells through AKT-mediated BAX activation. A genetic approach, through siRNA-mediated down-regulation of AKT, MCL-1, and BCL-xL, significantly decreased cell survival, demonstrating the importance of these cell survival factors for ABT-199 resistance. Our findings suggest a novel mechanism that modulates the expression and activity of pro-survival proteins to confer treatment resistance that could be exploited by a rational combination therapeutic regimen that could be effective for treating lymphoid malignancies.
Figures

Similar articles
-
Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism.Cancer Res. 2013 Feb 15;73(4):1340-51. doi: 10.1158/0008-5472.CAN-12-1365. Epub 2012 Dec 12. Cancer Res. 2013. PMID: 23243017 Free PMC article.
-
PI3K/mTOR dual inhibitor NVP-BEZ235 decreases Mcl-1 expression and sensitizes ovarian carcinoma cells to Bcl-xL-targeting strategies, provided that Bim expression is induced.Cancer Lett. 2014 Jun 28;348(1-2):38-49. doi: 10.1016/j.canlet.2014.03.001. Epub 2014 Mar 18. Cancer Lett. 2014. PMID: 24650799
-
Co-administration of the mTORC1/TORC2 inhibitor INK128 and the Bcl-2/Bcl-xL antagonist ABT-737 kills human myeloid leukemia cells through Mcl-1 down-regulation and AKT inactivation.Haematologica. 2015 Dec;100(12):1553-63. doi: 10.3324/haematol.2015.130351. Epub 2015 Oct 9. Haematologica. 2015. PMID: 26452980 Free PMC article.
-
Tipping the balance: toward rational combination therapies to overcome venetoclax resistance in mantle cell lymphoma.Leukemia. 2022 Sep;36(9):2165-2176. doi: 10.1038/s41375-022-01627-9. Epub 2022 Jun 20. Leukemia. 2022. PMID: 35725771 Free PMC article. Review.
-
Pathways and mechanisms of venetoclax resistance.Leuk Lymphoma. 2017 Sep;58(9):1-17. doi: 10.1080/10428194.2017.1283032. Epub 2017 Jan 31. Leuk Lymphoma. 2017. PMID: 28140720 Free PMC article. Review.
Cited by
-
AT101 [(-)-Gossypol] Selectively Inhibits MCL1 and Sensitizes Carcinoma to BH3 Mimetics by Inducing and Stabilizing NOXA.Cancers (Basel). 2020 Aug 15;12(8):2298. doi: 10.3390/cancers12082298. Cancers (Basel). 2020. PMID: 32824203 Free PMC article.
-
Venetoclax and dinaciclib elicit synergistic preclinical efficacy against hypodiploid acute lymphoblastic leukemia.Haematologica. 2023 May 1;108(5):1272-1283. doi: 10.3324/haematol.2022.281443. Haematologica. 2023. PMID: 36700399 Free PMC article.
-
Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes.Nat Commun. 2018 Sep 12;9(1):3694. doi: 10.1038/s41467-018-05984-x. Nat Commun. 2018. PMID: 30209285 Free PMC article.
-
Targeting Apoptosis in ALL.Curr Hematol Malig Rep. 2022 Apr;17(2):53-60. doi: 10.1007/s11899-022-00661-9. Epub 2022 May 11. Curr Hematol Malig Rep. 2022. PMID: 35538391 Review.
-
CDK5 Inhibitor Downregulates Mcl-1 and Sensitizes Pancreatic Cancer Cell Lines to Navitoclax.Mol Pharmacol. 2019 Oct;96(4):419-429. doi: 10.1124/mol.119.116855. Mol Pharmacol. 2019. PMID: 31467029 Free PMC article.
References
-
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–511. - PubMed
-
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002; 346: 1937–1947. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
